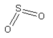
先决条件:需要先掌握利用Materials Visualizer绘制苯甲酰胺分子的教程研究背景:此教程基于Harrison等人(2006年)发表的一篇论文“Cu(111)和Ni(111)表面SO2和SO3结构的密度泛函理论研究”(Density functional theory investigation of the structure of SO2 and SO3 on Cu(111) and Ni (111))。研究SO2和SO3分子的吸附,是基于电站排放物中清除此类环境污染物的需求。本文对SO2分子吸附的几种几何构型进行了DFT计算。可以通过以下两种方式将Adsorption Locator模块作为初始构型准备和筛选工具:
单击“模块Modules”工具栏上的Adsorption Locator模块后的箭头,然后从下拉列表中选择计算Calculation以打开Adsorption Locator Calculation对话框,该过程中要确保Ni (1 1 1).xsd是活动文档,处于打开状态。在Setup选项卡上,设置精度Quality为Fine。从吸附质Adsorbate下拉列表中选择SO2 Forcite GeomOpt/SO2.xsd,将加载数量Loading设置为1。在Energy选项卡上,从“力场Forcefield”下拉列表中选择COMPASSIII。在Location选项卡上,选中“由原子集合定义的表面区域Surface region defined by atom set”复选框。当计算精度Quality设为Fine时,计算中将进行多次模拟退火循环,每个循环运行多步,以获得更精确的统计信息。(2)接下来选择原子,从而定义表面区域。
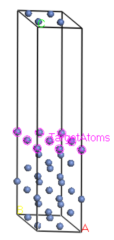
选择顶层的Ni原子,被选中的原子显示为黄色。
在Adsorption Locator Calculation对话框的Location选项卡中,单击Add按钮,从而将选定的原子包含在为吸附计算设置的目标原子集合TargetAtoms set中。选中“设置最大吸附距离Set maximum adsorption distance”复选框,输入设定值5.0。
参考文献:”Density functional theory investigation of the structure of SO2 and SO3 on Cu(111) and Ni(111)” by M. J. Harrison, D. P. Woodruff, and J. Robinson, Surface Science, 600, 1827, (2006).