-
开始
-
准备计算结构模型
-
定义原子配对
-
利用LST/QST/CG方法计算过渡态
-
过渡态优化

 ,从下拉列表中选择3D Atomistic Document。
,从下拉列表中选择3D Atomistic Document。 ,然后单击Clean按钮
,然后单击Clean按钮 。
。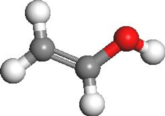
 。双击乙烯醇结构中的任何一个原子选中所有原子。
。双击乙烯醇结构中的任何一个原子选中所有原子。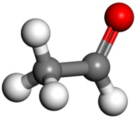
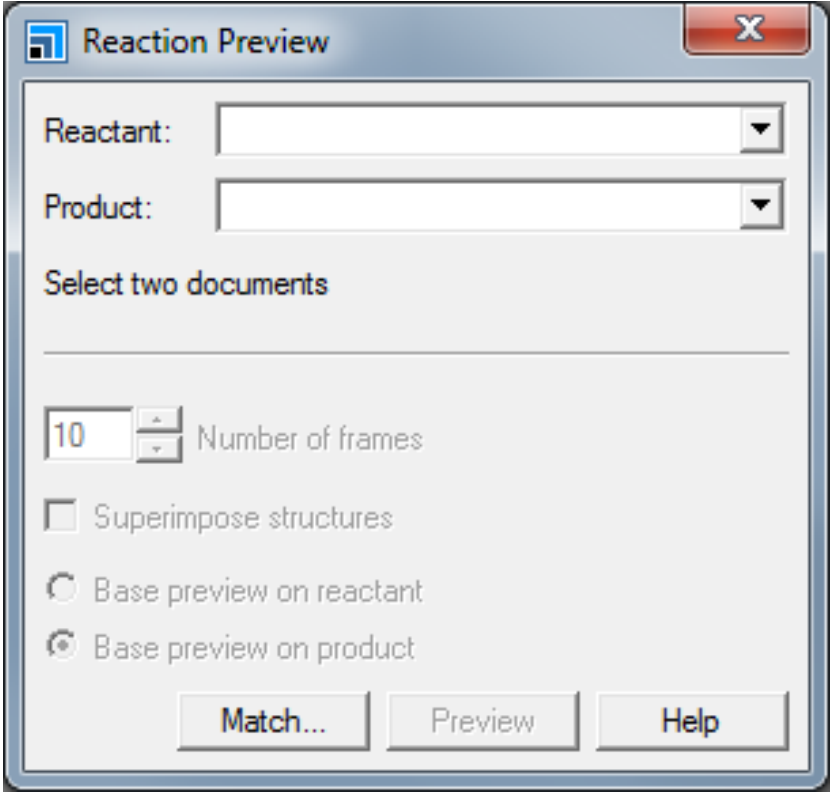
 。
。声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!


开始
准备计算结构模型
定义原子配对
利用LST/QST/CG方法计算过渡态
过渡态优化
,从下拉列表中选择3D Atomistic Document。,然后单击Clean按钮。
。双击乙烯醇结构中的任何一个原子选中所有原子。。