

总结:本文华算科技系统介绍了路易斯酸位点的定义、常见材料及其在异相催化中的作用机制。路易斯酸位点是能够接受电子对的活性中心,常见于金属氧化物、沸石分子筛及金属有机框架(MOF)等材料中。
文中结合多种催化反应实例,阐述了其在促进反应物吸附与活化、调控反应路径及抑制副反应等方面的重要作用,并通过实验与DFT计算揭示其结构-性能关系。这些认识为通过调控路易斯酸位点实现高效、选择性催化剂的设计提供了理论依据与实践指导。
路易斯酸位点(Lewis acid site)是指能够接受电子对的活性中心,它在异相催化中扮演着重要角色。与提供质子的Brønsted酸位不同,路易斯酸位点通常由缺电子的金属中心或空轨道构成,能够与底物的富电子部位配位,从而活化底物分子。
例如,在某些酶中,不参与氧化还原的金属离子(如酶中的Mg+或Ca2+)就充当路易斯酸,通过配位底物、稳定过渡态来降低活化能。在固体催化剂中,路易斯酸位常由未饱和配位的金属阳离子(如表面缺陷位的金属)或构型缺陷产生。
这些位点可以显著影响反应物的吸附与转化路径,是理解催化机制和设计新催化剂的关键。
常见的路易斯酸位点材料
金属氧化物中的路易斯酸位点
许多金属氧化物表面的金属中心由于配位不饱和而表现出路易斯酸性。例如,Al2O3、ZrO2、TiO2等氧化物表面的低配位金属离子可以接受电子对,是典型的路易斯酸中心。
CeO2等还具有同时提供路易斯酸/碱对的表面位,用于吸附极性分子如CO2。此外,新兴的单原子催化剂中,分散在碳载体上的金属单原子往往以M–N或M–O配位形式存在,也可视为“类”路易斯酸中心。
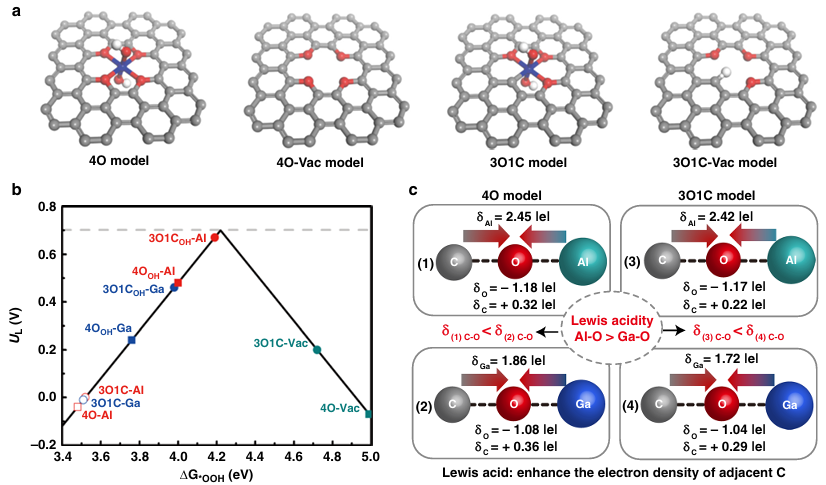
例如,有研究将氧化铝和氧化镓原子锚定在碳材料上作为Lewis酸位点,发现这些位点能极大调控相邻碳活性位的电子结构。DFT计算显示,八面体配位的Al–O或Ga–O单原子位具有强路易斯酸性,可影响临近碳上吸附物的键强度,从而优化反应中间体的吸附/解吸平衡。
这类原子级路易斯酸中心在电催化等领域展现出独特性能,证明缺陷金属位的路易斯酸性质对于催化活性具有重要意义。

DOI: 10.1038/s41467-020-19309-4
沸石分子筛中的路易斯酸位点
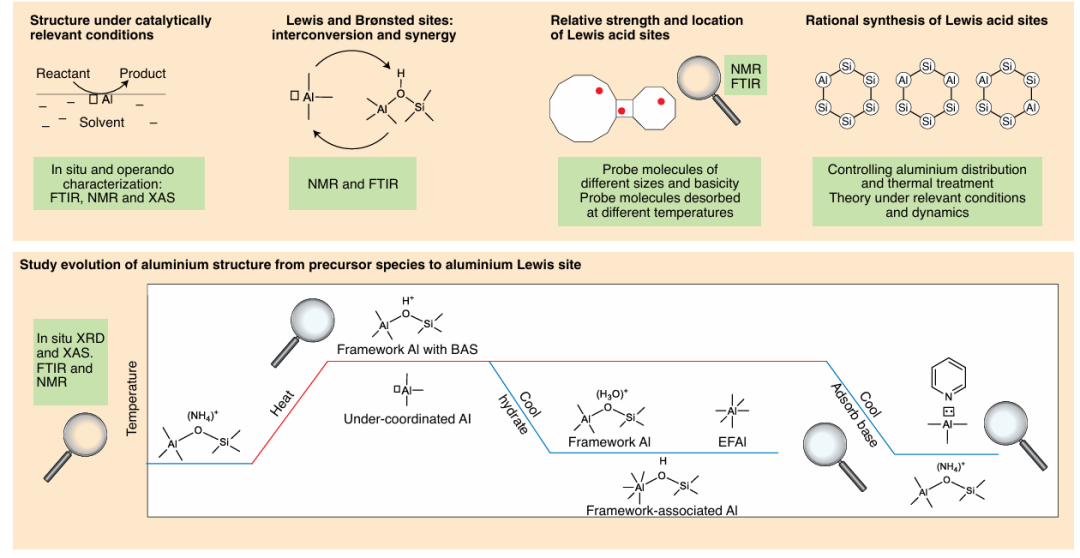
沸石是一类重要的固体酸催化剂,除典型的Brønsted酸(如框架Al-OH-Si)外,也包含路易斯酸位点。
沸石中的路易斯酸中心主要来源有三类:
(1)骨架内嵌杂原子:例如在硅铝分子筛中引入四价金属(Sn4+、Ti4+、Zr4+等)替代Si形成骨架位,因其周围缺少配位氧而成为Lewis酸中心;
(2)脱框架金属:高温水热处理等可使部分框架Al脱出形成骨架外的阳离子(如“脱铝”产生的Al3+簇),这些额外框架铝物种常表现为路易斯酸;
(3)缺陷相关位:如沸石框架内部缺少配位氧的铝位点等。这些路易斯酸Al位点的结构和作用长期存在争议,但公认它们在许多酸催化反应中发挥重要作用。值得注意的是,路易斯酸位点数量未必与脱框架铝含量简单正相关,不同表征手段需要结合才能准确确定其强度、位置和结构。
典型例子包括Sn-Beta分子筛,其中Sn4+取代框架Si形成强路易斯酸中心,已被广泛用于糖类转化等反应。Sn-Beta能够高效催化葡萄糖等醣类的异构或转化,例如在160°C下将葡萄糖转化为甲基乳酸,产率可达60%以上。
又如Y型沸石在脱铝后产生的Al3+簇也提供Lewis酸性,被认为可催化烃类的异构化和裂解反应。
DOI: 10.1038/s41563-020-0751-3
金属有机框架(MOF)中的路易斯酸位点
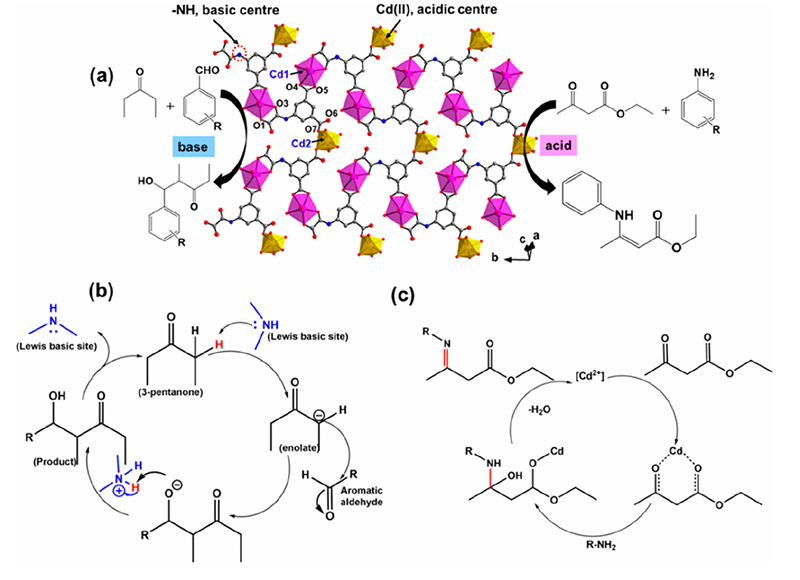
金属有机框架因其可设计的结构和功能多样性,成为构建路易斯酸位点的新平台。MOF中节点金属通常被有机配体配位包裹,但通过后合成活化(如去除框架中配位的溶剂或配体),许多MOF可生成配位不饱和金属位,表现出路易斯酸性质。
例如,经典的MIL-101(Cr)、UiO-66(Zr)等MOF在除去孔道客体分子后,其金属节点(Cr3+或Zr4+六核团簇)上会出现开放的金属配位位,这些位点能够与底物分子配位,从而催化多种有机反应。
MOF中的Lewis酸中心可以用于类似于传统路易斯酸催化剂的反应,例如Friedel-Crafts酰基化、烯烃碗式加成、环加成等。更有趣的是,通过简单调变合成条件或后修饰,研究者可以调控MOF节点的Lewis酸强度以及引入协同的碱性位点,实现双功能催化。
例如,有报告显示在Zr基MOF中引入含碱性基团的配体,可形成同时具备路易斯酸-路易斯碱对的“受阻路易斯酸碱”位点,用于活化小分子CO2。
DOI: 10.1021/acsorginorgau.3c00033
Lewis酸位点在异相催化反应中的作用
路易斯酸位点通过多种机制影响异相催化反应性能,主要体现在:促进底物吸附与活化、调控反应路径以及抑制副反应等方面。
吸附与活化反应物
路易斯酸位点最直接的作用是增强对反应物的吸附并活化反应键,从而降低反应能垒。由于Lewis酸可以接受配位电子对,它往往与底物分子中的富电子基团(如>C=O、-OH、环氧基、碳碳双键的π键等)发生配位,从而极化底物键、提高底物的反应活性。
DFT计算和实验研究均支持这一点:当底物与催化剂的路易斯酸中心配位时,其键长和电荷分布发生变化,使得键更易断裂或官能团更易受到亲电/亲核进攻。
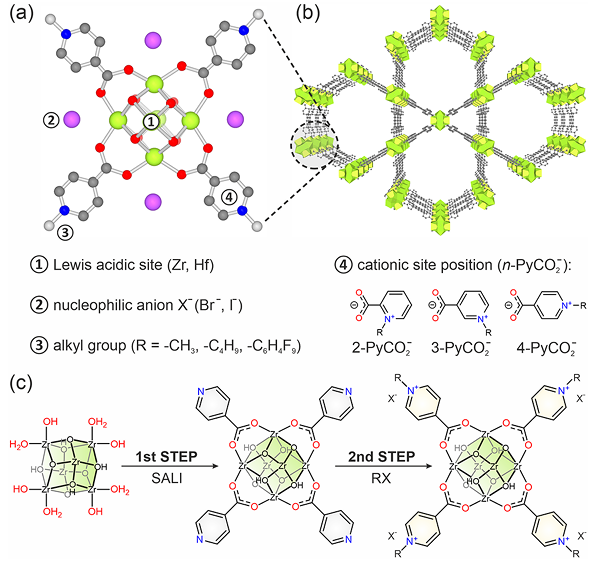
例如,在CO2环加成反应中,Zr基MOF提供的开放Zr4+位点作为路易斯酸,首先与环氧化物底物的氧原子配位,显著削弱了C–O键并增大环氧张力。
这一吸附复合物使环氧化物更易被亲核试剂(如卤阴离子或CO2本身)进攻开环,DFT机制分析进一步揭示了Zr–O配位降低了环氧开环的过渡态能垒。因此,无需高温高压,在室温常压下即可高效将CO2与环氧化物转化为碳酸酯产物。
DOI: 10.1021/acsami.0c20437
调控反应路径
除了促进单步反应外,路易斯酸位还能通过选择性稳定特定的中间体或过渡态,从而引导反应沿着特定路径进行。这一作用在多步反应网络和复杂反应中尤为重要,路易斯酸可以改变竞争反应的能垒高低,使主要产物的选择性提升。
一个突出的例子是甲醇制烃(MTH)反应中,沸石中的路易斯酸对反应机理的影响。H-ZSM-5等沸石传统上通过Brønsted酸位将甲醇转化为烯烃再进一步生成烃类。但研究发现,如果沸石中存在额外的Lewis酸(例如脱框架的Al物种或引入的Zn2+离子),反应路径和产物分布会发生明显变化。
实验-理论联合研究(JACS 2019)表明,Brønsted酸与Lewis酸在H-ZSM-5中存在协同作用,可促进甲醇转化初始C–C键的生成。
脱铝产生的框架外Al3+作为Lewis酸中心,可与沸石Brønsted酸位附近生成活性对,从而加速甲醇转化的诱导期,通过甲醛与甲醇偶联等“碳基成核”步骤更快地形成第一分子烯烃。
核磁共振和DFT计算检测到了甲醇在Lewis酸Al上形成的表面甲氧基和羟基中间体,这种物种被认为引发了一条不同于纯Brønsted酸机制的新“羰基途径”,降低了成碳-碳键的能垒。
另一面,在甲醇制芳烃(MTA)过程中,引入Zn等路易斯酸中心能够显著调整产物选择性。Zn2+阳离子交换到HZSM-5后,一方面弱化了Brønsted酸强度,另一方面提供了新的Lewis酸位用于烃类中间体的脱氢芳构化。
研究表明,随着Zn含量增加,催化剂的芳构化活性中心增多,烯烃更倾向于在Lewis酸位上脱氢生成芳香烃,而不是发生Brønsted酸催化的氢转移生成烷烃。
DFT分析支持这一点:Zn2+可以稳定芳香环形成所需的碳正离子过渡态,并抑制氢转移过渡态,从而偏向芳香烃路径、抑制副产烷烃。实际结果是,含Zn的催化剂芳烃收率大幅提高,而烷烃和焦炭前驱物明显减少。
由此可见,通过引入或调整路易斯酸位点,催化剂可以稳定特定中间体或改变关键步骤的速控步,从而引导反应沿期望路径进行,实现对产物分布的调控。
DOI: 10.1016/j.fmre.2021.08.002
抑制副反应
路易斯酸位点另一个重要作用是抑制不良副反应,从而提高目标产物的选择性和催化剂稳定性。在许多酸催化过程中,副反应往往由强Brønsted酸位引发,例如过度的脱水、聚合导致积碳等。
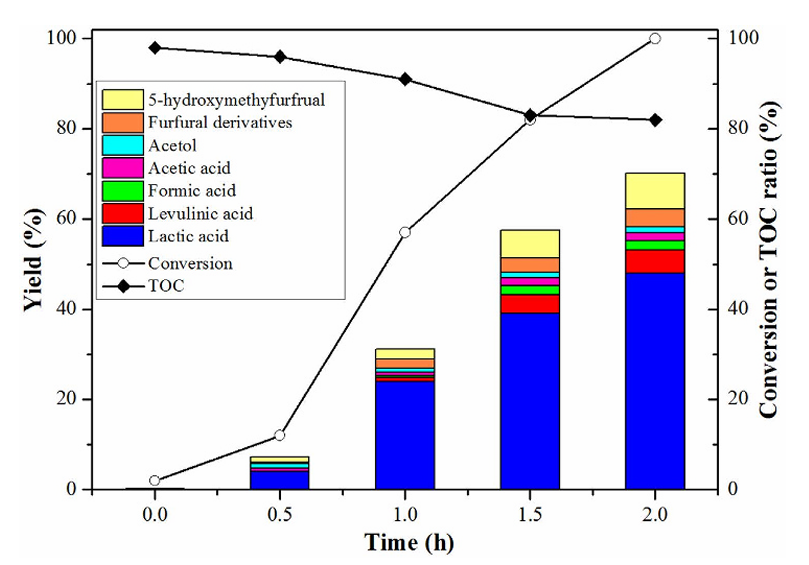
而路易斯酸由于不提供质子,在很多情况下可以避免这些副反应,或者通过与其他位点协同来抑制副产物形成。一个典型事例是糖制乳酸反应中副产5-羟甲基糠醛(HMF)的问题。
纯Sn-Beta沸石在水溶液中将糖类转化为乳酸时,生成的乳酸等有机酸本身会作为Brønsted酸催化剂,导致果糖进一步脱水成HMF等副产物,严重降低乳酸收率。
为了解决这一难题,研究者引入Zn改性来构筑Lewis酸-碱双功能催化剂:通过固态离子交换将Zn引入Sn-Beta,得到Zn-Sn-Beta催化剂。结果显示,在无碱条件的水溶液中,Zn-Sn-Beta可以将蔗糖完全转化,在2小时内乳酸收率达到54%,远高于未改性Sn-Beta。
机理研究表明,Zn的引入一方面增强了路易斯酸位数量,另一方面在Sn-Beta中产生了碱性位点(如Zn相关的氧物种)。这些碱性位可以中和反应过程中生成的有机酸,避免其作为Brønsted酸催化进一步的脱水副反应;同时,Lewis酸位Sn4+继续负责主反应(糖异构和重排形成乳酸)。
换句话说,Zn-Sn-Beta通过酸碱协同抑制了HMF等副产物的生成,使主产物乳酸选择性大大提高。这一策略体现了调控路易斯酸/碱平衡来抑制副反应的有效性。少副反应发生,这是提高选择性和催化剂寿命的重要策略。
DOI: 10.1038/srep26713