
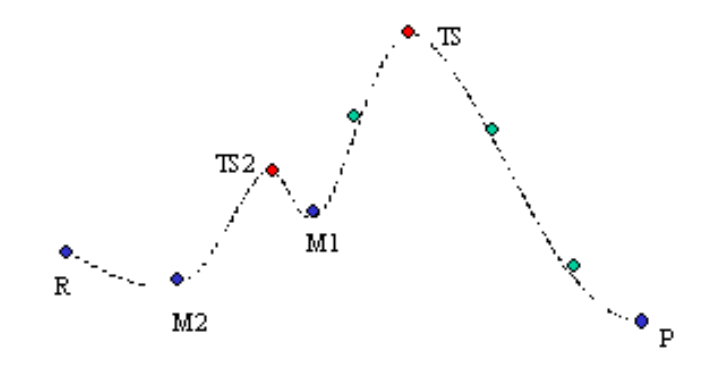

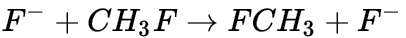
-
开始 -
准备计算结构模型 -
准备过渡态计算的输入轨迹文档 -
使用LST/QST计算过渡态 -
使用TS确认执行NEB计算
 ,从下拉列表中选择3D Atomistic Document。
,从下拉列表中选择3D Atomistic Document。 。将其中一个氢原子更改为氟原子。
。将其中一个氢原子更改为氟原子。 。
。 ,选择Calculation,或从菜单栏中选择Modules | DMol3 | Calculation。
,选择Calculation,或从菜单栏中选择Modules | DMol3 | Calculation。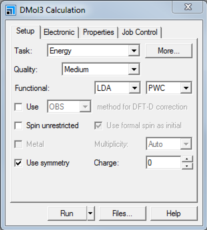
 ,从下拉列表中选择Periodic Table…,打开Periodic Table对话框。
,从下拉列表中选择Periodic Table…,打开Periodic Table对话框。 的下拉箭头,然后从下拉列表中选择Distance。选择新创建的C-F键,按住鼠标左键并拖动鼠标,直到距离增加到约3 Å。
的下拉箭头,然后从下拉列表中选择Distance。选择新创建的C-F键,按住鼠标左键并拖动鼠标,直到距离增加到约3 Å。 。按下CTRL键并单击上一步创建的距离测量,然后单击拉长的C-F键以将其选中。按下DELETE键。
。按下CTRL键并单击上一步创建的距离测量,然后单击拉长的C-F键以将其选中。按下DELETE键。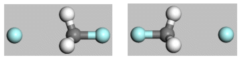
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!