-
开始 -
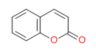
建立和优化输入结构模型 -
在气相中计算香豆素光谱 -
在水存在的条件下计算香豆素光谱 -
激发态的几何构型优化

 ,选择Calculation,打开DMol3 Calculation对话框。
,选择Calculation,打开DMol3 Calculation对话框。 ,选择Analysis,打开DMol3 Analysis对话框。
,选择Analysis,打开DMol3 Analysis对话框。声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!
 ,选择Calculation,打开DMol3 Calculation对话框。,选择Analysis,打开DMol3 Analysis对话框。
,选择Calculation,打开DMol3 Calculation对话框。,选择Analysis,打开DMol3 Analysis对话框。