


本文华算科技介绍了原位同步辐射XAS的原理、主要区域、装置及电催化反应机制中对反应中间体、氧空位等研究。
XAS理论始于1920年,Friche和Hertz首次观察到X射线吸收谱在吸收边处呈现精细结构,随后该技术引起物理学家的广泛关注。
1971年,Sayers、Stern和Lytle基于单电子单次散射理论,提出X射线能量与被测材料结构之间的关系,并给出被广泛接受的表达式。结合傅里叶变换,将几何结构与XAS特征清晰关联,使XAS成为表征材料的利器。不久,同步辐射光源的X射线引入测试,大幅提高了数据采集速度和精度。
经过40年的发展,EXAFS的理论、实验及数据分析已趋完善。图1展示了X射线吸收光谱(XAS)测量的示意图,其中包括同步加速器储存环、单色器、样品和探测器。
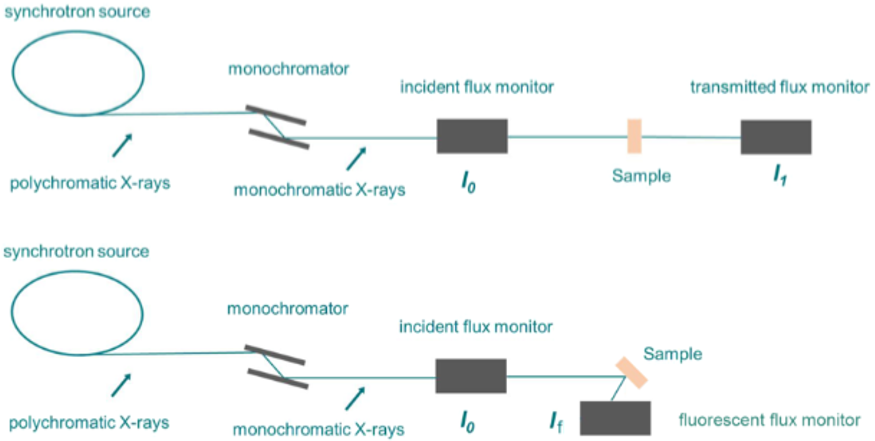
图1:传透射模式和荧光模式用于测量XAS。DOI:10.1002/smsc.202100023
X射线吸收光谱(XAS)是一种强大的光谱技术,能够在原子和分子水平上揭示物质的结构和化学态变化。
XAS光谱主要包括三个区域:前边缘(pre-edge)、X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)。
前边缘区域对中心原子周围的低对称环境和未占据电子态敏感;XANES区域可提供有关吸收原子的氧化态、配位对称性以及配体的性质等信息;EXAFS区域则主要用于确定吸收原子周围的原子种类、配位数以及原子间距等结构参数。
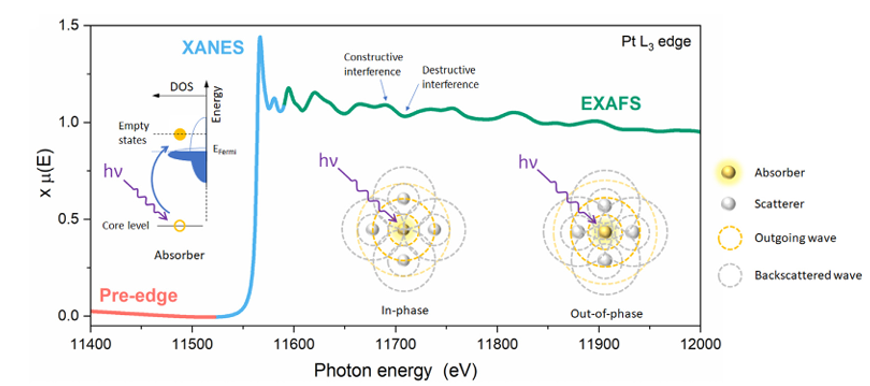
图2:XAS光谱图包含三个区域:起始边缘区(红色)、XANES区(青色)和EXAFS区(绿色)。DOI:10.1016/j.coelec.2020.100681
尽管原位XAS可用于研究催化剂的电子和几何结构,但探究催化反应的机制是不可行的,特别是无法实时监测金属催化中心的动态变化。原位X射线吸收光谱(XAS)借助同步辐射的高能量与穿透力,可在原子尺度解析电催化反应中催化剂的电子和几何演变。
在过去十年中,原位XAS已被用于系统地研究电催化水分解和二氧化碳还原反应的机制。用于电化学测试的原位XAS仪器发展迅速,主要由X射线光谱仪和独特的电化学电池组成。
仪器的最新发展集中在电化学电池及组件设计上,电池包含主体、进出液管道、窗口、平台/电池固定器(图3a)。在水解反应中,电化学电池可按透射和荧光模式操作(图3b、c)。
透射模式要求样品薄且催化剂含量多,X射线能够穿透并被收集(图3b),装置有Si3N4制的X射线透明窗口,催化剂沉积在窗口的金/导电层上作为工作电极,窗口两侧分别对着电解质和射线源,其及导电层的厚度会影响X射线的衰减。
荧光模式与催化剂的厚度和数量无关,元素灵敏度更高,更适合实际反应(图3c)。在CO2还原反应(CO2RR)中,使用原位XAS流动电池,包含CO2气体室和液体室,由阴离子交换膜分隔,通过气体扩散电极隔开。
还使用了MEA系统,可减少欧姆损耗。通过利用原位XAS(原位X射线吸收光谱)设备,可以详细追踪电催化过程中催化剂表面的动态变化。
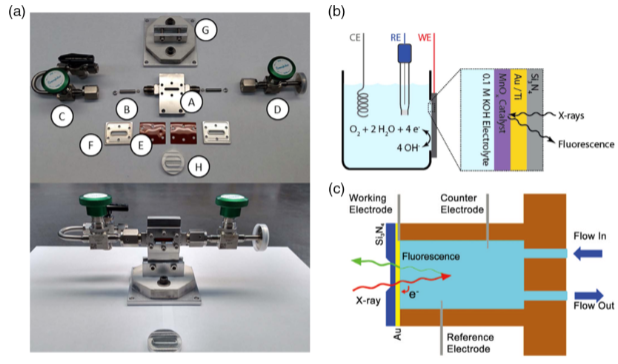
图3:原位XAS仪器图
原位XAS通常通过分析近边缘位置的变化来识别活性位点,这一变化表明了某种变化的发生。在电催化过程中,金属中心与配位原子之间的键长存在极化状态和偏差。
通常,电催化剂中的目标原子充当催化中心,而其他配位原子则在改变相关原子的性质或为吸附物提供适宜环境方面发挥辅助作用。
1、反应中间体:研究员通过光诱导晶格应变策略合成了具有晶格应变的NiFe金属有机框架(MOF),用于双功能氧电催化。
通过原位同步辐射傅里叶变换红外(SR-FTIR)结合软X射线吸收光谱(XAS)技术,他们对活性成分和反应中间体进行了探测。如图4所示,同位素标记的原位SR-FTIR结果证实,在1048 cm-1处的振动峰归因于在相对于可逆氢电极(RHE)施加1.6 V电位时表面中间体超氧阴离子OOH*。
原位软X射线吸收光谱显示出875.8 eV的峰值,这归因于随着施加电位增加至相对于RHE 1.6 V,Ni4+物质的存在。当施加的电压切换回相对于参考电极(RHE)为1.2伏的初始电压时,Ni4+物质便消失了。
与晶格应变的NiFe金属有机框架(MOF)相比,原始的NiFe MOF在施加电位高达相对于RHE的1.7 V时,875.8 eV处并未显示出任何特征峰。
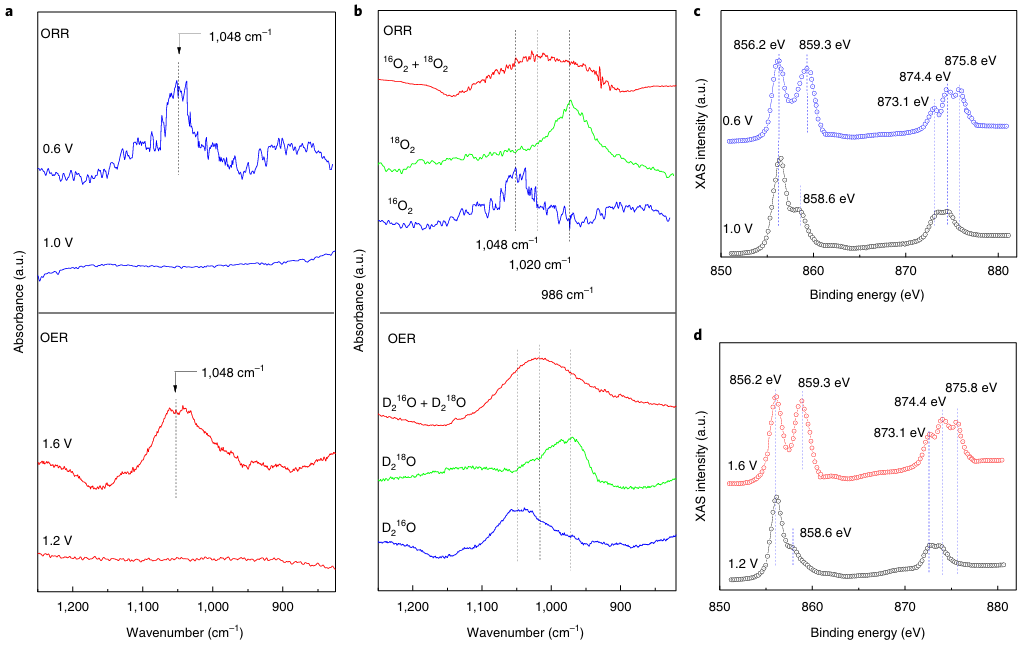
图4:在氧还原反应(ORR)和氧析出反应(OER)过程中,会形成超氧中间体以及高价态的Ni4+物质。DOI:10.1002/smsc.202100023
2、氧空位位点:研究员通过氩等离子体构建了一种富含氧空位的Co3O4作为模型催化剂,利用原位XAS研究了在氧析出反应(OER)过程中氧空位位点的动态演变。
为了保持电中性,氧空位会导致Co3O4中形成低价态的钴。原位Co K-edge XAS通过监测吸收边能量随电位升高而正向位移,实时捕捉到氧空位促使Co2+在更低电位预氧化为高价态;
同步记录的EXAFS显示Co-O配位数在1.45 V先增后降,定量证明氧空位被OH–优先填充并快速转化为Co-OOH,其随后去质子化释放O2,从而直接确立了氧空位驱动的动态重构机理。
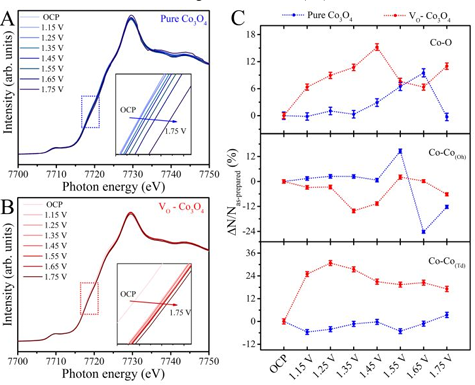
图5:原位XAFS Co K-edge结果。DOI:10.1021/jacs.0c00257
3、原子界面位点:研究员通过界面工程开发了一种碳/氮固定化的Mn单一催化剂用于氧电催化。
通过原位XAS技术,作者们发现,距离伸展的Mn2+-N2C2原子界面位点在氧还原反应(ORR)中起到了催化作用,而距离收缩的Mn4+-N2C2部分则在氧析出反应(OER)中充当了活性中心。
Mn K边XANES光谱显示,对工作电极施加一系列正电位后,XANES边沿向更高能量方向移动,这表明锰的价态有所增加。EXAFS光谱表明,对于OER来说,Mn-N/C的峰值位置从1.47 Å显著向低R方向移动至1.43 Å。
理论计算证实,与其他三种结构相比,固定一个锰原子的两个氮原子和两个碳原子的几何结构是最稳定的。
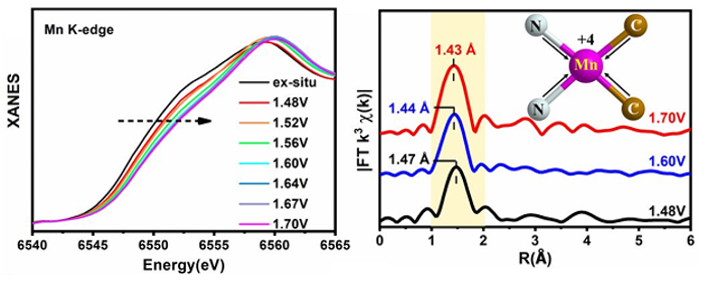
图6:锰硫酸盐的Operando XAFS特性分析。DOI:10.1021/acs.nanolett.0c01925
4、前沿轨道:通过原位软X射线吸收光谱可以研究前沿轨道(过渡金属阳离子的nd轨道和C/N/O阴离子的2p轨道)。整个实验必须在超高真空环境中进行,以避免低能入射软 X 射线的不必要损失。
原位O K边XAS光谱被分为三个区域:1)能量范围为525 – 534 eV,对应于从O 1s到O 2p的M-13d电子跃迁过程;2)534 – 540 eV区域,表现出水分子的近边缘特征;3)540 eV以上区域,显示O K边的扩展X射线吸收精细结构(EXAFS)。
研究员利用原位OK边XAS揭示了电沉积NiFeOxHy OER电催化剂中O阴离子的局部键合状态和对称性特征。
如图7所示,529、529.9、531.1和532.5 eV的四个峰被识别为从O 1s到O 2p与Ni 3d t2g、Fe 3d t2g、Oπ*(O2气体的)以及Fe 3d eg的杂化跃迁,分别发生在OER电位为1.78 V时(由于镍价态的增加而出现),并在还原电位为1.18 V时可逆地后退。
该峰的变化与O 2p和Ni 3d之间的杂化变化有关,这种变化是由从O位点向Ni位点的电子注入或提取所导致的(Ni3+-O²⁻ ↔ Ni(3-δ)+-O(2-δ)-)。
相比之下,529.9 eV处的峰(O 1s – O 2p/Fe 3d t2g)和532.4 eV处的峰(O 1s – O ₂p/Fe 3d e2g)随着施加电压的变化而略有变化,这表明与Ni位点相比,Fe位点的变化很小。
原位OK边揭示了O ₂p与过渡金属离子的轨道杂化以及它们之间的电荷转移,这揭示了电催化过程中前线轨道的演变。由于吸附中间体引起的金属氮–碳电催化剂的价态变化和电荷转移也可以通过原位软XAS来确认。
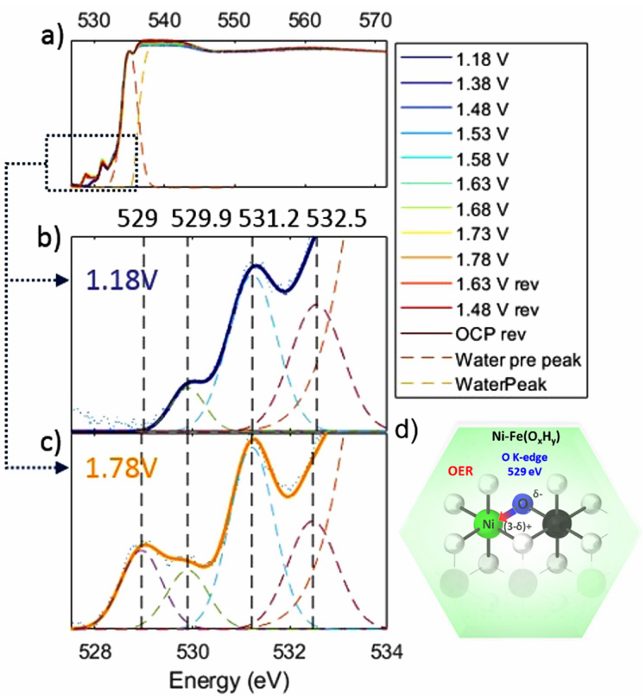
图7:镍铁催化剂的原位XAS O K边缘光谱。DOI:10.1038/s41598-018-37307-x
5、单原子活性位点:金属氮-碳电催化剂的价态变化和由于吸附中间体引起的电荷转移也可以通过原位软X射线吸收光谱得到证实。
研究员通过将氮固定在磷化碳氮(PCN)框架中,构建了原子分散的钴催化剂,用于碱性析氢反应(HER)。使用原位XAFS确定了钴单原子催化剂活性位点的动态结构。
如图8所示,研究人员在碱性析氢反应过程中实时捕捉到Co1/PCN单原子催化剂活性位点的动态演化:XANES显示电位由开路向-0.1 V负移时,吸收边连续升高约0.9 eV,定量拟合表明Co氧化态由+2.02升至+2.40,意味着OH⁻吸附形成高价HO–Co1–N2活性中心;
EXAFS仅出现1.5 – 1.7 Å单主峰且无Co-Co信号,证实始终为单原子状态,同时该峰位从1.63 Å(Co–N)低-R移至1.56 Å并出现新的Co–O配位,拟合结果显示配位环境由初始4 N演化为“2 N + 1 O”再到“2 N + 2 O”,对应HO⁻吸附后再结合H2O生成H₂O–(HO–Co1–N2)反应中间体,从而直接锁定高价HO–Co1–N2为碱性HER的真正活性位点。
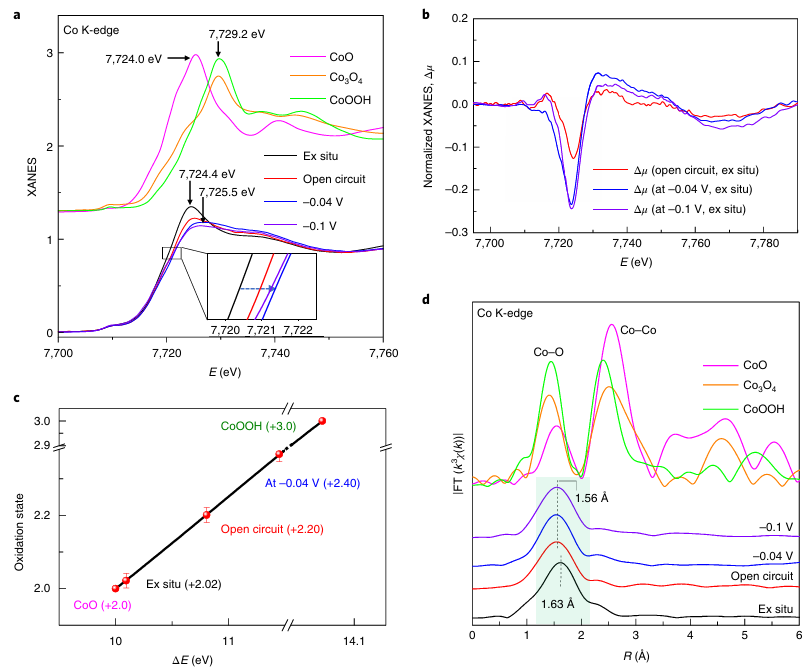
图8:Co L边XANES光谱。DOI:10.1038/s41929-018-0203-5
本文介绍了原位同步辐射XAS的原理、主要区域、装置及在电催化水分解中对反应中间体、氧空位位点、原子界面位点等的研究,展望其未来在提升时间分辨率、结合其他技术深入解析电催化机制等方面潜力,助力高效电催化剂研发。