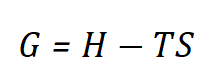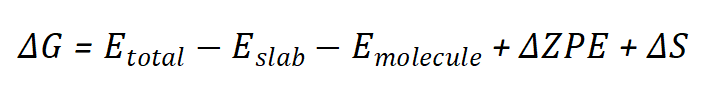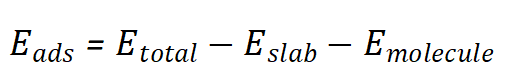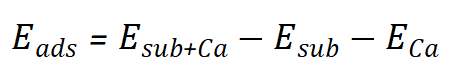在材料科学、催化反应、电化学和表面化学等领域,自由能和吸附能是描述物质相互作用、能量变化和反应路径的重要物理量。
它们不仅决定了材料的稳定性、催化活性和反应速率,还为催化剂设计、材料筛选和界面工程提供了理论依据。以下华算科技将从自由能的定义与计算、吸附能的定义与计算、自由能与吸附能的关系、实际应用中的分析方法、实验与模拟的结合等方面进行详细分析。
自由能(Free Energy)是热力学中描述系统在恒温恒压条件下能量状态的重要参数。在电化学和催化反应中,通常使用吉布斯自由能(Gibbs Free Energy, G)来描述反应的热力学可行性。其定义为:
其中,H是系统的焓,T是温度,S是系统的熵。吉布斯自由能的变化ΔG决定了反应是否自发进行。在催化反应中,ΔG的变化可以反映反应路径的难易程度,从而判断催化剂的活性和选择性。
在理论计算中,自由能通常通过密度泛函理论(DFT)结合分子动力学模拟或量子化学计算来获得。常用的计算方法包括:
静态计算:通过优化结构并计算能量,结合零点振动能(ZPE)和熵变(ΔS)来修正自由能。
动态计算:通过分子动力学模拟(MD)或路径积分方法(PIMD)来考虑温度和振动对自由能的影响。
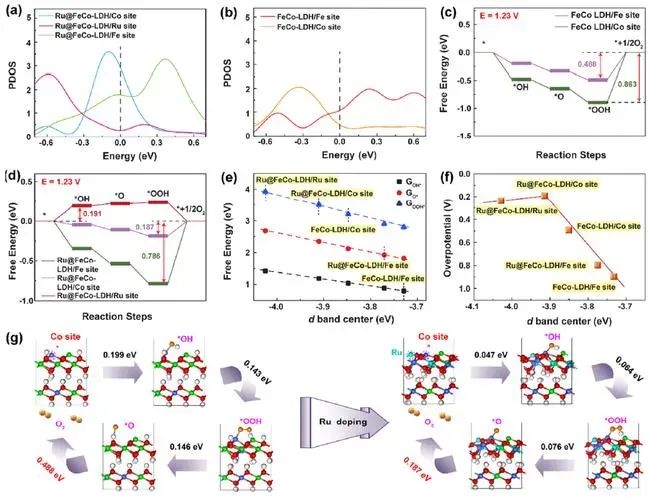
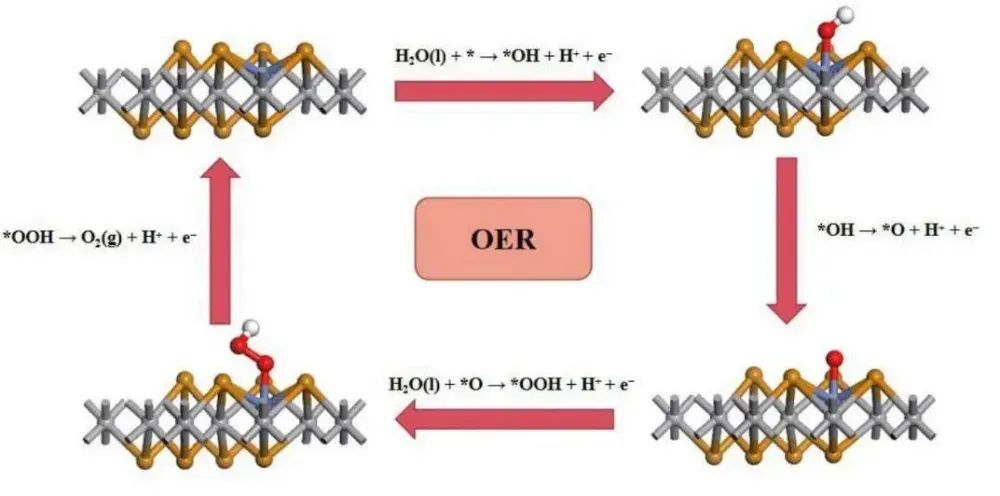
在ORR(氧气还原反应)中,自由能的变化通常用于构建自由能台阶图(Free Energy Diagram),以揭示反应路径中的能量变化。
吸附能(Adsorption Energy)是描述分子或原子在固体表面吸附时释放的能量。其定义为:
吸附能的正负值表示吸附过程是放热还是吸热。吸附能越大(绝对值越小),吸附越稳定。
吸附能的计算通常基于DFT计算,结合赝势方法(Pseudopotential)和平面波基组(Plane Wave Basis Set)进行。常用的软件包括VASP、Quantum ESPRESSO、Materials Studio等。
结构优化:对吸附体系进行几何优化,确保达到能量最低点。
基态能量计算:计算催化剂表面Eslab和反应物分子Emolecule的能量。
在白藜芦醇对钙的吸附能计算中,作者通过DFT计算了白藜芦醇(C14H12O3)对钙(Ca)的吸附能。计算中使用了PBE泛函和PAW赝势方法,并考虑了DFT-D3 Grime方法进行范德华修正。吸附能的计算公式为:
其中,Esub+Ca是吸附体系的总能量,Esub是白藜芦醇表面的总能量,ECa是钙原子的孤立能量。吸附能为负值,表示吸附过程是放热的,吸附越稳定。
自由能与吸附能之间存在密切关系。吸附能是自由能计算的基础之一,而自由能则进一步考虑了温度、熵和溶剂化效应等因素。在催化反应中,吸附能决定了反应物在催化剂表面的稳定性,而自由能则决定了反应路径的可行性。
在ORR反应中,吸附能决定了反应物在催化剂表面的稳定性,而自由能则决定了反应路径的能垒。例如,在ORR中,O₂分子的吸附能决定了其在催化剂表面的稳定性,而自由能则决定了O₂解离为OOH的能垒。
自由能台阶图是分析催化反应路径的重要工具。通过计算各中间体的吸附能和自由能,可以绘制出反应路径中的能量变化,从而识别决速步骤(RDS)和催化活性位点。
计算自由能:结合零点振动能和熵变,计算各中间体的自由能。
绘制自由能台阶图:将各中间体的自由能数据输入绘图软件(如Origin、VASP等),绘制自由能变化曲线。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!