

说明:金属–载体相互作用(MSI)指负载型催化剂中金属与载体的物理化学作用,含电子、几何及化学键合效应。可通过密度泛函理论(DFT)、分子动力学(MD)模拟,分析态密度、吸附能等参数。其研究为设计高效催化剂提供理论基础,推动能源催化等领域技术革新。
金属和载体的相互作用是什么?
金属–载体相互作用(MSI)是负载型催化剂中金属纳米颗粒(NPs)或单原子(SAs)与载体间物理化学作用的统称,通过电子转移、几何重构及化学键合等机制调控催化活性与选择性。
具体而言,经典强金属–载体相互作用(SMSI)表现为载体氧化物向金属表面迁移形成包覆层(如TiO₂包裹Pt NPs),这种包覆层一方面通过空间位阻抑制金属团聚,另一方面改变金属表面电子态,例如使d带中心下移从而优化反应物吸附能。
电子金属 – 载体相互作用(EMSI)则侧重载体与金属间的电荷重新分布,典型如石墨烯负载的金属催化剂中,碳载体的π电子与金属d轨道杂化导致电荷转移,进而调节金属的d带中心位置及对CO、H₂等分子的吸附强度,这种电子耦合效应在电催化析氢反应中可显著降低反应能垒。
此外,根据载体性质(可还原/不可还原氧化物),MSI还呈现氧化型与还原型差异:在Au/ZnO体系中,发生“氧化型SMSI”,电子从Au纳米颗粒转移至ZnO载体,形成Au⁺-O⁻界面物种,增强对CO的吸附活化能力;而在Pt/TiO₂体系中,还原气氛下TiO₂释放氧空位,Ti³⁺向Pt转移电子,构成 “还原型SMSI”,提升对NOₓ的催化还原活性。
这些相互作用机制通过X射线光电子能谱(XPS)、原位透射电镜(in-situ TEM)及密度泛函理论(DFT)计算得以验证——DFT可量化载体对金属d带电子结构的调制程度。
MSI的深入研究为设计高效催化剂提供了理论基础,例如通过调控载体氧空位浓度增强EMSI,或利用可还原氧化物载体诱导SMSI包覆层以提升催化剂抗中毒能力,推动了能源催化、环境治理等领域的技术革新。

DOI:10.3390/molecules28052262
MSI的核心作用机制
电子效应
在金属–载体相互作用(MSI)的电子效应机制中,d带中心理论揭示了载体调控金属催化性能的核心路径:密度泛函理论(DFT)计算证实,载体与金属间的电荷转移会显著偏移金属的d带中心(εd),进而定量调节反应中间体的吸附强度。
以Pt/CN(碳氮化物)体系为例,CN载体诱导Pt的d带中心下移,弱化了含氧物种(如 OH*)在Pt表面的吸附能,通过优化氧还原反应(ORR)的决速步能垒提升催化活性;而在硫掺杂碳载体负载的Pt纳米簇体系中,出现罕见的反向电子转移(Pt→C),形成富电子Pt位点,通过增强H * 吸附能显著提升析氢反应(HER)动力学。
界面电子重分布机制则在电子金属–载体相互作用(EMSI)模型中得以阐释:载体表面的电荷极化(如氧化物载体的氧空位诱导)使金属纳米颗粒(NPs)外表面形成电荷梯度,这种远程电荷效应直接影响金属位点的功函数——例如TiO₂载体的氧空位使邻近Pt NPs的功函数降低0.2 eV,通过改变电子逸出能力调节反应物分子的吸附解离路径,该效应在CO氧化等涉及多步电子转移的反应中尤为显著。
DFT计算进一步表明,界面电子重分布可使金属表面的d带电子局域化程度提升15%-20%,为实验中观察到的催化活性增强提供了电子结构层面的理论支撑。
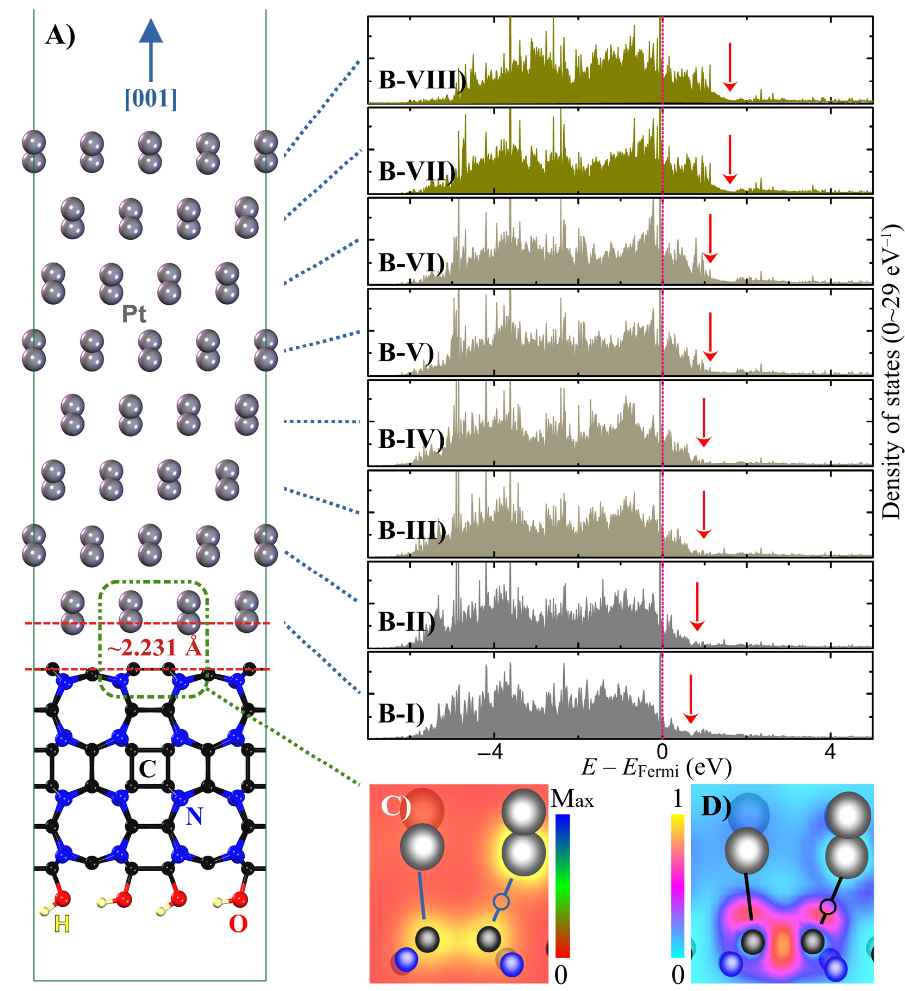
DOI:10.3390/nano14110921
几何效应
在金属–载体相互作用(MSI)的几何效应机制中,界面原子配位重构与金属颗粒尺寸效应通过改变活性位点的局部几何环境调控催化性能。
基于密度泛函理论(DFT)模拟,Cu/MgO界面处的氧原子因同时键合MgO晶格中的Mg²⁺与金属Cu的表面Cu原子,形成独特的双配位结构,导致其对反应分子的吸附能显著增加(约0.5 eV),而远离界面的Cu位点吸附能仅变化0.1 eV,体现了界面区域原子配位环境对吸附行为的特异性影响。
这种配位重构通过改变活性位点的配位数与键长,直接影响反应中间体的吸附构型与活化能垒,在CO氧化等涉及氧物种活化的反应中至关重要。
金属颗粒尺寸效应则表现为载体对金属吸附性能的调制强度随颗粒尺寸变化呈现临界行为:当Cu纳米颗粒(NPs)尺寸小于2 nm时,MgO载体诱导的吸附能偏移可达1 eV,根源在于小尺寸颗粒表面原子占比高,界面原子的配位不饱和性显著增强;而当颗粒尺寸超过2.5 nm后,内部体相原子占主导,载体引起的几何效应逐渐消失,吸附能偏移趋于稳定。
DFT计算进一步揭示,尺寸效应本质是界面原子比例与配位环境变化的协同作用——小尺寸颗粒中界面原子占比超过30%,导致表面原子平均配位数从体相的12降至8-9,形成更多低配位活性位点,其对CO、H₂等分子的吸附能较体相位点变化可达0.8 eV。
这种尺寸依赖的几何效应在纳米催化中具有重要意义,例如通过调控金属NPs尺寸至临界值以下,可最大化载体对活性位点几何结构的调制作用,实现对C-C偶联、加氢反应等的选择性优化。
几何效应的研究结合原位电镜表征与理论计算,为设计具有特定配位环境与尺寸的高效负载型催化剂提供了原子尺度的调控策略。
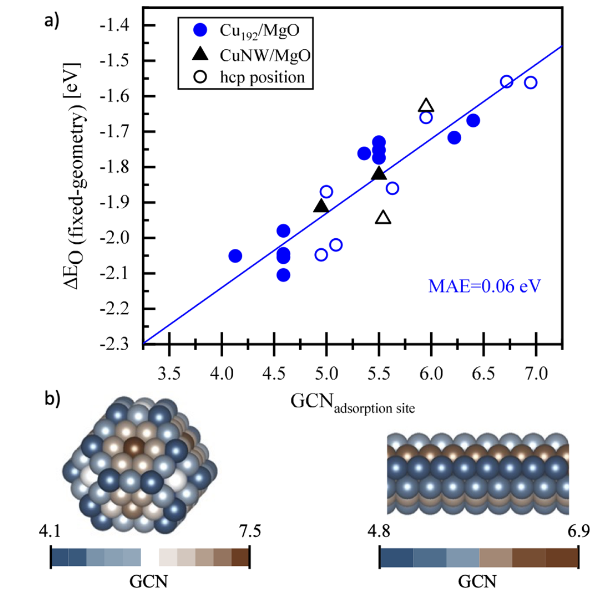
DOI:10.1021/acsomega.3c00502
化学键合效应
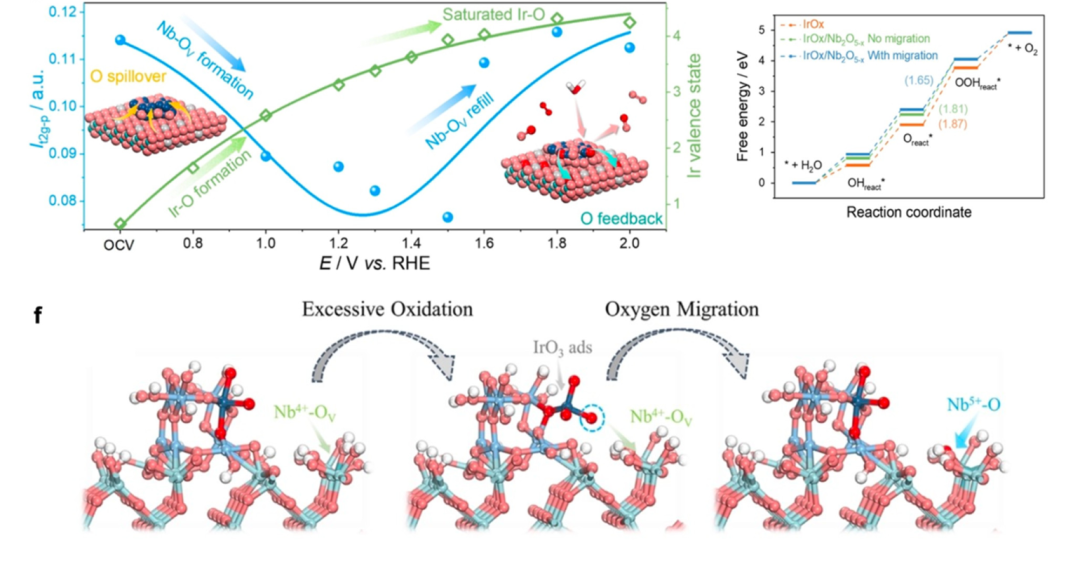
在金属–载体相互作用(MSI)的化学键合效应中,金属–氧键的形成与载体缺陷锚定通过强化学作用调控金属活性位点的结构与稳定性。
在金属/氧化物界面体系(如Pt-TiO₂)中,密度泛函理论(DFT)计算证实,界面区域形成具有共价特性的Pt-O-Ti键,该键通过共享电子对连接金属Pt与氧化物载体TiO₂,一方面通过强相互作用抑制金属纳米颗粒(NPs)的团聚,使Pt NPs在高温反应中保持高分散状态;另一方面促进载体氧空位的生成——Pt-O-Ti 键的电子离域效应导致TiO₂晶格中氧原子的结合能降低0.3 eV,使其更易脱离晶格形成氧空位,为CO吸附活化等反应提供活性位点。
载体缺陷锚定机制则体现为可还原氧化物(如CeO₂)表面的氧空位对金属原子(如Rh单原子)的特异性捕获:CeO₂晶格中的氧空位因局部正电荷富集(Ce³⁺位点),与金属原子形成强配位键(如Rh-O键),通过化学键合能(约1.2 eV)将Rh原子稳定锚定在载体表面,避免其迁移团聚。
这种锚定效应不仅提升金属物种的分散度(如Rh在CeO₂表面的单原子负载量可达1.5 wt%),更通过界面化学键的电子耦合改变金属的电子结构——例如Rh-O键的形成使Rh的d带中心上移0.2 eV,增强对NOₓ等分子的吸附能力,在氮氧化物还原反应中显著提升催化效率。
化学键合效应的本质是金属与载体通过形成共价键、配位键等强相互作用,实现对金属位点的几何结构(如配位数、键长)与电子结构(如d带电子分布)的双重调控。
DFT计算进一步揭示,金属–氧键的键长(如Pt-O键长约2.05 Å)与键级(约0.8)直接影响界面电荷转移程度,而载体缺陷锚定的键合能与缺陷类型(如CeO₂的氧空位、ZrO₂的晶界缺陷)密切相关。
这些机制为设计高稳定性、高活性负载型催化剂提供了关键策略,例如通过调控载体氧空位浓度优化金属–氧键强度,或利用缺陷工程实现单原子催化剂的高效锚定,推动了多相催化在能源转化与环境治理中的实际应用。
DOI:10.1016/j.ese.2024.100443
MSI的理论模拟方法
密度泛函理论
在金属–载体相互作用(MSI)的理论模拟中,密度泛函理论(DFT)通过量子力学第一性原理计算,为揭示MSI的微观机制提供了核心工具。
其应用场景涵盖吸附能、态密度(DOS)、d带中心及Bader电荷转移量等关键参数的定量分析:通过计算反应物分子在金属–载体界面的吸附能,可确定活性位点的催化反应路径;态密度与d带中心分析则直接关联金属电子结构变化,例如载体诱导的d带中心偏移对中间体吸附强度的调控;Bader电荷分析可量化金属与载体间的电荷转移量,为电子金属–载体相互作用(EMSI)提供数据支撑。
针对单原子催化剂(SACs),DFT通过金属–氧结合能、载体带隙等描述符,预测金属单原子与载体的键合强度,例如在Fe-N-C单原子催化剂中,计算Fe-N₄活性位点的结合能以优化载体配体环境。
典型案例研究以Cu/MgO体系为例,DFT计算对比了Cu不同晶面(如密排面Cu (111) 与高指数面Cu (321))及纳米颗粒尺寸(如Cu₁₃团簇与大于2 nm 的Cu颗粒)的O吸附能,发现界面处Cu原子因同时配位MgO的Mg²⁺与吸附氧物种,其O吸附能较体相Cu位点高出0.5 eV以上,证实界面区域是MSI的主要贡献区。
进一步分析表明,小尺寸Cu纳米颗粒()因表面原子占比高,界面配位重构效应显著,导致吸附能偏移随尺寸减小呈指数增长,而当颗粒尺寸超过2.5 nm时,体相原子主导使尺寸效应趋于饱和。
这些计算结果与原位 X 射线吸收光谱(XAS)表征高度吻合,揭示了界面位点在CO氧化等反应中的关键作用。
DFT的优势在于能够从原子尺度解析MSI的电子效应、几何效应与化学键合效应,例如通过计算Pt-TiO₂界面的Pt-O-Ti键共价特性,阐释载体对金属纳米颗粒的稳定机制及氧空位生成动力学。
其输出的微观参数(如d带中心、键长、电荷密度)不仅为实验表征提供理论验证,更可作为催化剂设计的描述符,推动 “理论计算–实验验证” 循环在多相催化中的高效迭代。
随着计算效率提升与复杂模型(如周期性slab模型、溶剂化效应)的发展,DFT正从简化界面体系模拟向真实反应条件下的多尺度MSI研究拓展,成为连接微观结构与宏观催化性能的核心理论桥梁。
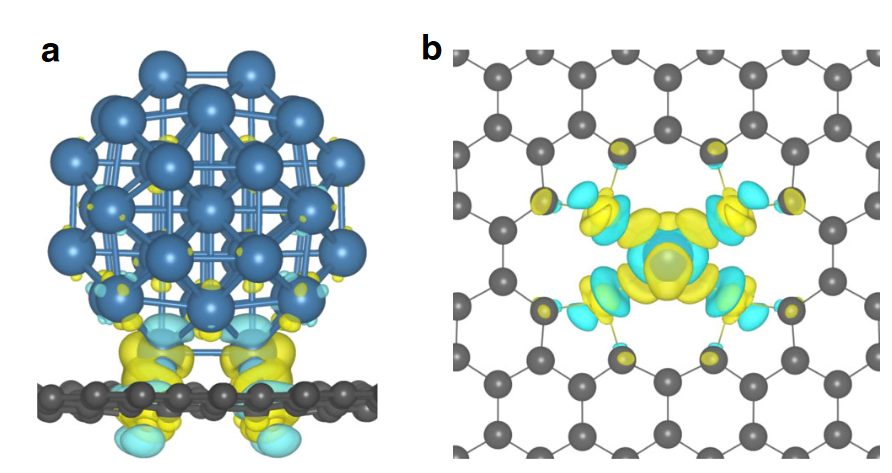
DOI:10.1038/s41467-019-12851-w
分子动力学(MD)模拟
在金属–载体相互作用(MSI)的分子动力学(MD)模拟研究中,力场选择与模拟流程设计是揭示界面动态行为的关键技术环节。
力场方面,嵌入原子势(EAM)通过考虑原子间的电子密度嵌入效应,精准描述金属键与离子键的协同作用,适用于金属/金属氧化物界面的原子扩散、颗粒团聚等动力学过程模拟,例如在Pt/TiO₂体系中,EAM力场可定量追踪高温下Pt纳米颗粒在载体表面的迁移与团聚路径。
而ReaxFF反应力场则通过键级–电荷相互作用模型,有效捕捉化学键的断裂与形成事件,尤其适用于涉及氧空位迁移、载体表面重构的MSI体系——如在CeO₂负载Rh单原子催化剂中,ReaxFF力场可模拟氧空位诱导的Rh-O键动态重组,揭示载体缺陷对金属位点稳定性的影响机制。
标准模拟流程遵循严格的热力学框架:首先基于实验或DFT优化结果初始化原子位置与速度,通过共轭梯度法进行能量最小化以消除不合理的原子间相互作用,随后在NVT或NpT系综下通过牛顿动力学方程积分追踪原子轨迹(时间步长通常设为1-2 fs),实时记录体系能量、原子坐标等数据。
模拟输出的轨迹文件经傅里叶变换、径向分布函数分析等处理,可定量表征界面原子扩散系数(如MgO载体表面Cu原子的扩散能垒为0.6 eV)、配位结构演化(如TiO₂氧空位附近Pt原子的配位数从5增至7)及颗粒尺寸变化(如高温下Au NPs从3 nm团聚至8 nm的动力学路径)。
这些动态信息与DFT静态计算形成互补,例如MD模拟揭示的Pt纳米颗粒在TiO₂表面的 “跳跃式” 迁移行为,为DFT计算提供了真实的界面结构初始构型,二者结合可更准确预测MSI对CO氧化反应能垒的调制效应(约降低0.3 eV)。
MD模拟的优势在于能够动态呈现MSI中界面原子的协同运动,如载体表面氧原子的热振动如何影响金属颗粒的电子结构,或剪切力作用下金属–载体界面的几何重构过程。
通过合理选择力场(如EAM的金属键描述优势与ReaxFF的反应活性优势),MD可在纳秒时间尺度上模拟实际催化反应中的高温、高压条件,为理解MSI在动态反应环境中的演化规律(如催化剂失活过程中的颗粒烧结、载体相变)提供关键数据支撑,推动多相催化体系从静态结构表征向动态机制解析的深度发展。

典型理论计算解析
态密度(DOS)
态密度(DOS)分析作为揭示金属–载体相互作用(MSI)中电子结构变化的核心工具,通过量化能级分布与电子占据状态,为阐释MSI的催化调控机制提供了电子尺度的理论支撑。
在Pt/CN(碳氮化物)体系中,DFT计算显示载体诱导的Pt-d带中心向下偏移约0.5 eV,这一电子结构重构通过弱化氧物种(O*)在Pt表面的吸附能,优化了氧还原反应(ORR)的决速步能垒,从而提升催化活性。
该现象的本质是CN载体的π电子与Pt-d轨道形成共轭相互作用,导致d带电子离域化程度增强,电子结构向低能态偏移。
而在Sc-Ti₂CO₂体系中,DOS分析揭示Sc-3d轨道与载体O-2p轨道发生显著杂化,形成宽化的杂化能带,这种轨道耦合效应使Sc位点对CO、NO等气体分子的吸附能增强约0.4 eV,根源在于杂化轨道的电子云重叠提升了金属–分子间的成键能力。
通过DOS谱图的分峰拟合可进一步量化轨道贡献——Sc-3d与O-2p的杂化强度在费米能级附近达35%,直接关联气体分子的吸附活化效率。
DOS分析的优势在于将MSI诱导的电子结构变化(如d带中心位移、轨道杂化程度)与催化性能(吸附能、反应能垒)建立定量关联,例如Pt-d带中心每下移0.1 eV,O*吸附能约降低0.06 eV,为设计高活性催化剂提供了可量化的电子结构描述符,推动了多相催化从经验设计向理论指导的精准调控发展。
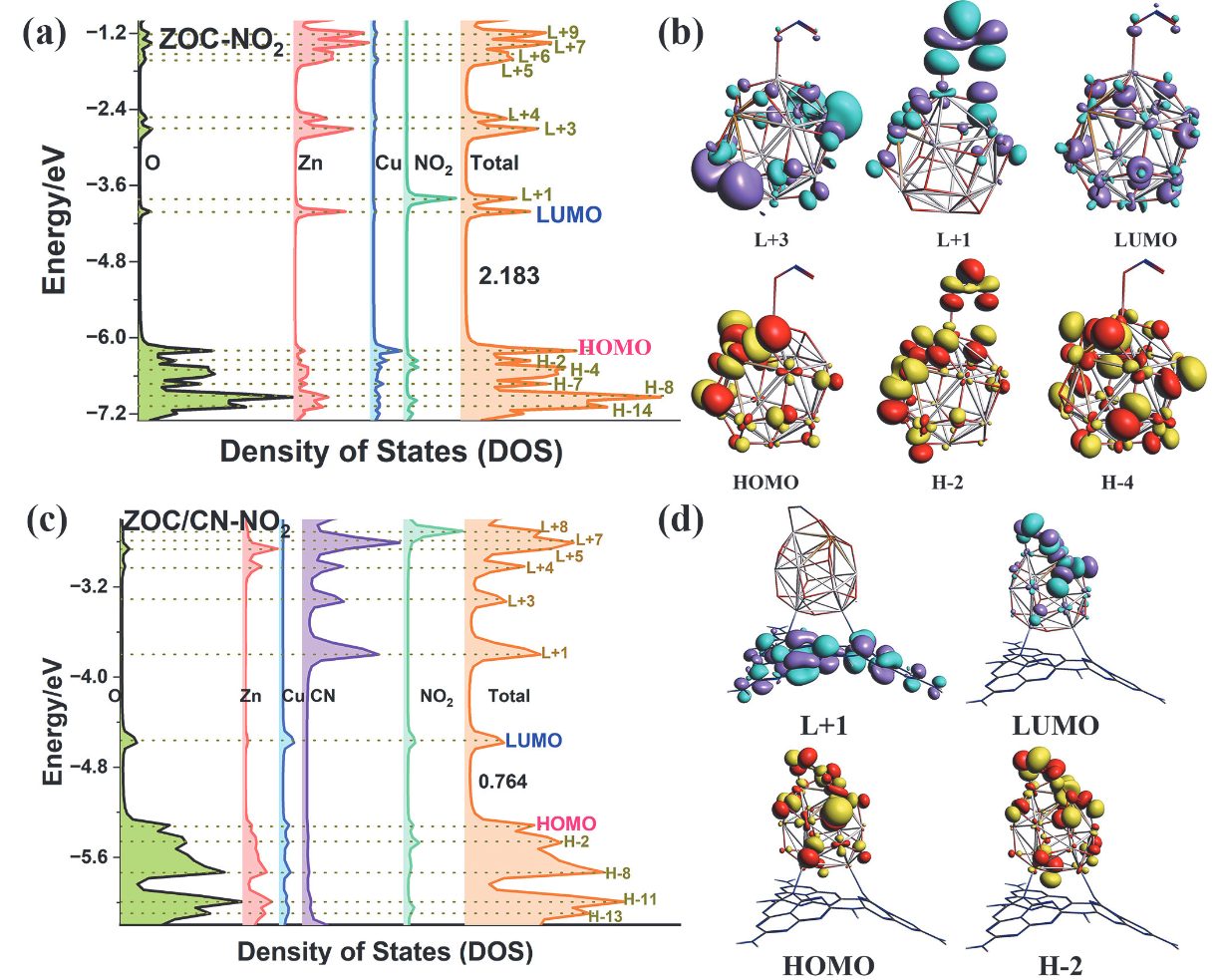
DOI:10.6023/A23060312
吸附能
在金属–载体相互作用(MSI)的吸附能研究中,界面位点特异性与尺寸效应共同构成调控催化活性的核心机制。
基于密度泛函理论(DFT)计算,Cu/MgO界面处的氧吸附能较非界面位点高出0.5 eV,根源在于界面氧原子同时配位MgO晶格中的Mg²⁺与金属Cu表面原子,形成独特的双配位结构,导致其电子云分布与成键环境发生显著重构,这种配位环境的改变使吸附能通过键能叠加效应得以提升。
尺寸效应则表现为Cu纳米颗粒(NPs)的氧吸附能随粒径减小呈现非线性增长——当颗粒尺寸从2.5 nm降至0.5 nm时,吸附能偏移达1 eV,本质是小尺寸颗粒表面原子占比激增(从20%升至70%),表面原子的配位不饱和性增强,形成更多低配位活性位点。
DFT进一步揭示,2.5 nm颗粒的表面原子平均配位数为9,而0.5 nm颗粒降至6,配位缺失导致d带电子离域化程度提升,使氧分子吸附时的轨道杂化强度增加0.4 eV。
这些吸附能的差异化分布直接影响催化反应路径:界面位点的高吸附能有利于氧物种活化,而小尺寸颗粒的强吸附效应可加速反应中间体转化,二者共同为设计高活性MSI催化剂提供了原子尺度的调控策略,例如通过调控载体界面结构或金属颗粒尺寸优化CO氧化、加氢等反应的能垒分布。
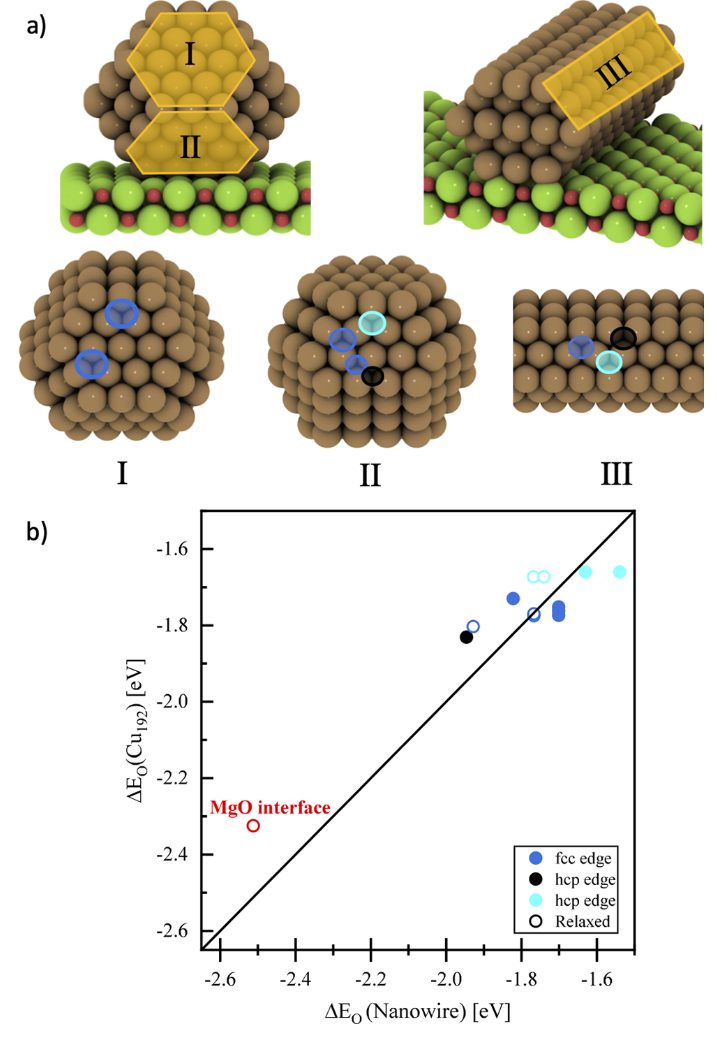
DOI:10.1021/acsomega.3c00502
Cu/MgO体系的DFT研究
Hakimioun等在《ACS Catalysis》发表的研究中,针对Cu/MgO体系的金属–载体相互作用(MSI)开展了系统性密度泛函理论(DFT)研究,通过构建多尺度计算模型与精细电子结构分析,揭示了界面效应与尺寸效应对催化活性位点的调控机制。
研究首先建立了不同原子数(13~200原子,对应粒径约0.5~3 nm)的Cu纳米颗粒(NPs)负载于MgO (100) 表面的模型,针对大尺寸颗粒计算成本高的问题,引入 “纳米线模型” 进行简化 —— 该模型通过周期性重复Cu纳米线结构模拟无限大颗粒的界面行为,在保持界面原子配位环境真实性的同时大幅降低计算量。
计算采用PBE泛函描述交换关联能,结合Bader电荷分析、局域态密度(DOS)及吸附能计算,从电子结构与几何结构双重维度解析MSI的微观本质。
关键结论表明,界面效应是Cu/MgO体系MSI的主导机制:仅当氧原子同时键合Cu NPs表面原子与MgO晶格中的Mg²⁺(即界面位点)时,其吸附能显著增加(ΔE=0.5 eV),而远离界面的Cu位点吸附能变化可忽略。
这种特异性源于界面氧原子的双配位结构——其同时参与Cu的金属键与MgO的离子键,导致电子云重新分布:Bader电荷分析显示,界面氧原子从Cu获得0.2 e⁻,从Mg²⁺获得0.1 e⁻,电荷富集使O 2p轨道能级下移,与Cu 3d轨道的杂化强度增强18%,最终表现为吸附能的协同升高。
局域DOS进一步证实,界面位点的Cu 3d带中心较体相位点上移0.15 eV,增强了对氧物种的轨道相互作用,这与传统观点中载体导致金属d带中心下移的电子效应形成差异,凸显了界面几何配位对电子结构的调制作用。
尺寸效应研究发现,Cu NPs的MSI强度存在明确的阈值行为:当颗粒尺寸大于2.5 nm时,吸附能偏移小于0.1 eV,MSI可忽略;而当尺寸减小至2 nm以下,吸附能随粒径减小呈指数增长,13原子Cu团簇(~0.8 nm)的界面位点吸附能较2.5 nm颗粒高出1 eV。
该现象的本质是表面原子占比与配位环境的协同变化——2.5 nm颗粒的表面原子占比为25%,界面原子仅占表面原子的30%,而0.8 nm团簇的表面原子占比达70%,界面原子占比提升至60%,导致平均配位数从体相的12降至7.5,低配位位点的比例显著增加。
DFT计算显示,低配位Cu原子的d轨道电子局域化程度更高,对O 2p轨道的吸引力增强,从而形成更强的化学吸附。这一结论为实验中观察到的小尺寸Cu NPs催化CO氧化活性增强提供了理论解释,表明通过调控颗粒尺寸至阈值以下()可最大化界面效应。
载体还原性对比研究表明,MgO作为不可还原氧化物,其MSI强度弱于可还原载体(如CeO₂),后者需额外考虑氧空位介导的电子转移效应。在MgO体系中,Cu与载体的相互作用主要限于界面配位重构,缺乏氧空位形成所需的晶格氧活化能力(MgO 的氧结合能为-5.2 eV,远高于CeO₂的-3.8 eV),因此电荷转移量仅为0.3 e⁻/ 界面O原子,显著低于CeO₂体系的0.6 e⁻/O 空位。
这种差异导致MgO载体对Cu电子结构的调制局限于界面区域,而CeO₂可通过体相氧空位扩散实现对金属颗粒整体电子态的调控(如d带中心下移0.3 eV)。
该发现为催化剂载体的理性选择提供了依据:不可还原载体适用于需要界面特异性吸附的反应(如CO₂吸附活化),而可还原载体更适合涉及多步电子转移的加氢或氧化反应。
研究方法的创新点在于“纳米线模型” 的引入,其通过结构周期性简化,将大尺寸颗粒的计算成本降低60%以上,同时保持界面原子配位环境的准确性(与全原子模型的吸附能计算偏差)。
结合Bader电荷与DOS的联合分析,该研究建立了从几何配位到电子结构再到吸附性能的完整关联链条,揭示了MSI中几何效应与电子效应的协同机制——界面配位重构是触发电子结构变化的几何基础,而电子结构的调整反过来影响吸附键的强度与性质。
该工作的科学意义在于,首次通过DFT量化了不可还原载体界面位点的特异性吸附效应,并明确了MSI尺寸效应的临界条件,为纳米催化剂的尺寸设计提供了精确的理论阈值(2.5 nm)。
此外,与可还原载体的对比研究,深化了对不同类型MSI(如经典SMSI与EMSI)作用机制的理解,推动了负载型催化剂设计从 “试错法” 向 “参数化调控” 的转变。
未来研究可进一步结合分子动力学模拟,探索高温反应条件下界面结构的动态演化,以及引入实验表征(如原位X射线吸收光谱)验证理论预测,从而构建更贴近实际催化环境的MSI模型。
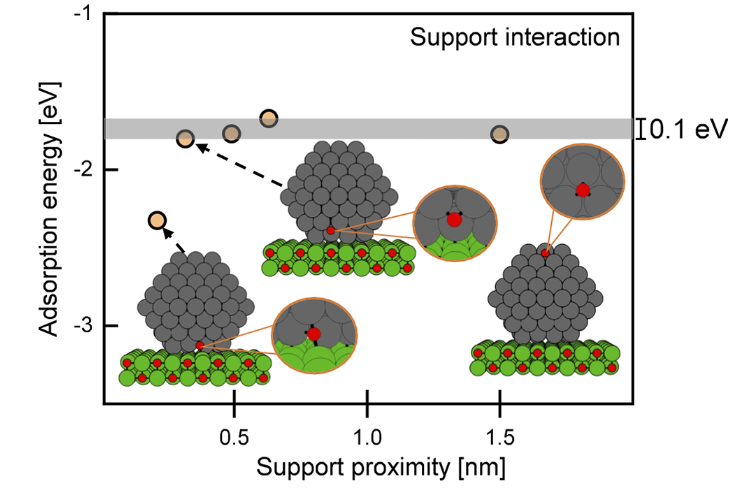
DOI:10.1021/acsomega.3c00502
总结
在金属–载体相互作用(MSI)的前沿研究中,多尺度模拟通过整合密度泛函理论(DFT)与分子动力学(MD),构建了从电子结构到动态演化的跨尺度研究框架——DFT从量子力学层面解析界面电子转移与化学键合(如Pt-O-Ti共价键形成),MD则追踪高温下原子扩散与颗粒烧结的动力学过程(如Cu NPs在MgO表面的迁移),二者协同揭示MSI在反应条件下的动态演化机制。
机器学习辅助技术通过遗传编程算法,将DFT计算的微观参数(如金属–氧结合能、载体带隙)转化为MSI描述符,建立预测单原子催化剂(SACs)稳定性的定量模型,例如通过训练神经网络势函数(NNP)快速评估Rh/CeO₂体系中氧空位对金属–载体键合强度的影响,将计算效率提升3个数量级。
复杂环境建模则聚焦电催化体系中溶剂化效应与外电场对MSI的调控,通过构建含explicit溶剂的周期性slab模型,DFT计算揭示水合离子在金属–载体界面的吸附重构了电子分布(如改变Pt的d带中心偏移量0.2 eV),而外电场作用下的非平衡态模拟进一步表明,界面偶极层可调制金属–载体电荷转移量达40%,为理解燃料电池阴极MSI的电位依赖性提供了理论基础。
这些方法的融合推动MSI研究从理想界面模型向真实反应环境跨越,为设计高稳定性、高活性催化体系提供了多维度的理论支撑。