

说明:表面是材料外1-3层原子层,因不饱和键致结构和性质异于体相。可从几何结构和功能特性分类,DFT通过Slab模型等研究表面,如过渡金属掺杂Cu (111) 催化案例,助于理解表面作用机制。
什么是表面?




表面是材料与外界(气体、液体、真空等)接触的最外层几层的原子层,其原子因处于最外层而缺失相邻原子,形成不饱和键,这使其几何结构、电子态和化学性质与材料内部(体相)显著不同。
具体而言,表面原子为降低自身能量,会在垂直方向发生位移(弛豫)或在水平方向进行重排(重构);其电子态常出现表面态、悬挂键等局域态。
这种独特性让表面在材料的光电效应、催化反应等过程中扮演着关键角色,例如表面的结构变化和特殊电子态会直接影响材料对光的吸收、电子的传输以及与反应物的作用效率等,深入理解表面的这些特性,能让我们更好地认识材料与外界环境相互作用的奥秘,为材料性能的优化和新功能的开发提供重要依据。
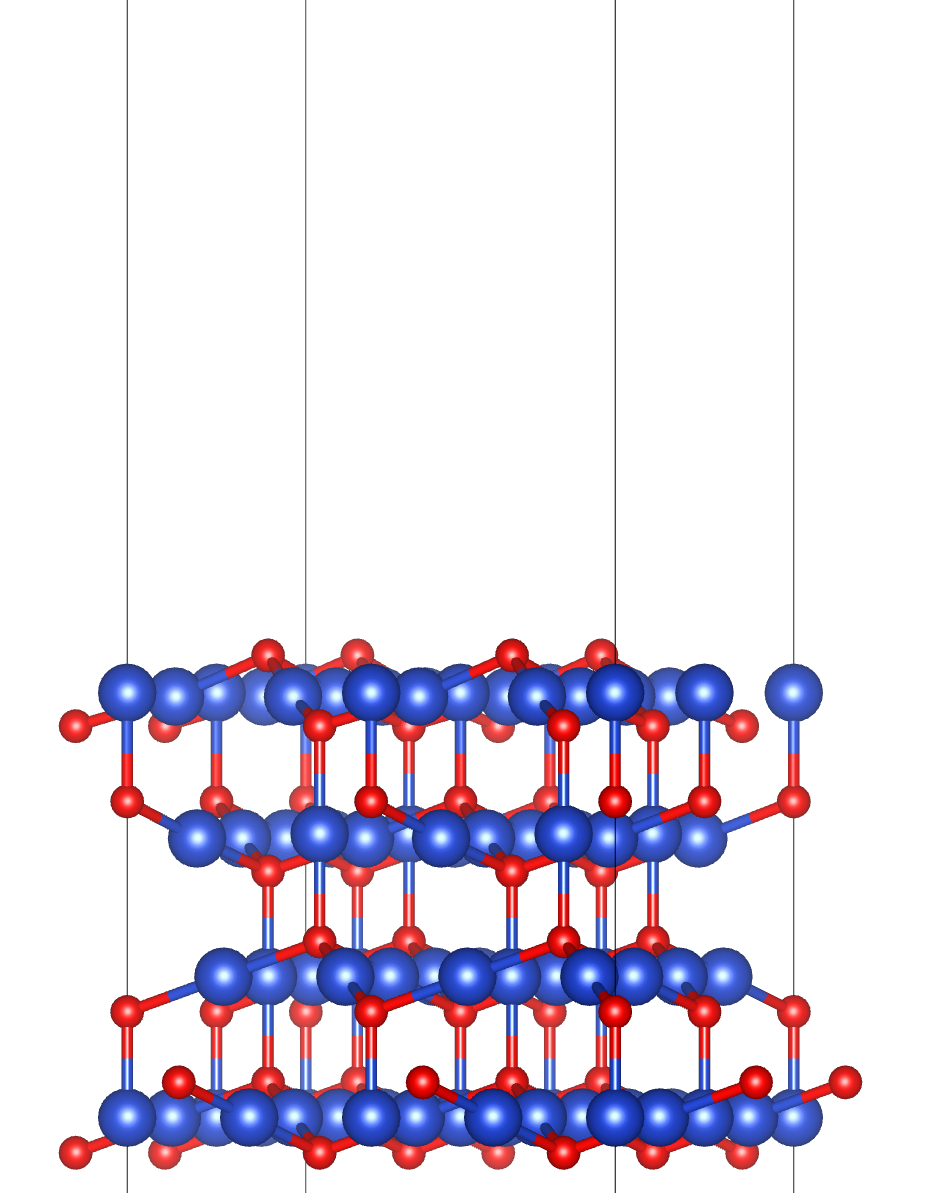
表面的分类
表面的分类可从几何结构与功能特性两个维度展开。
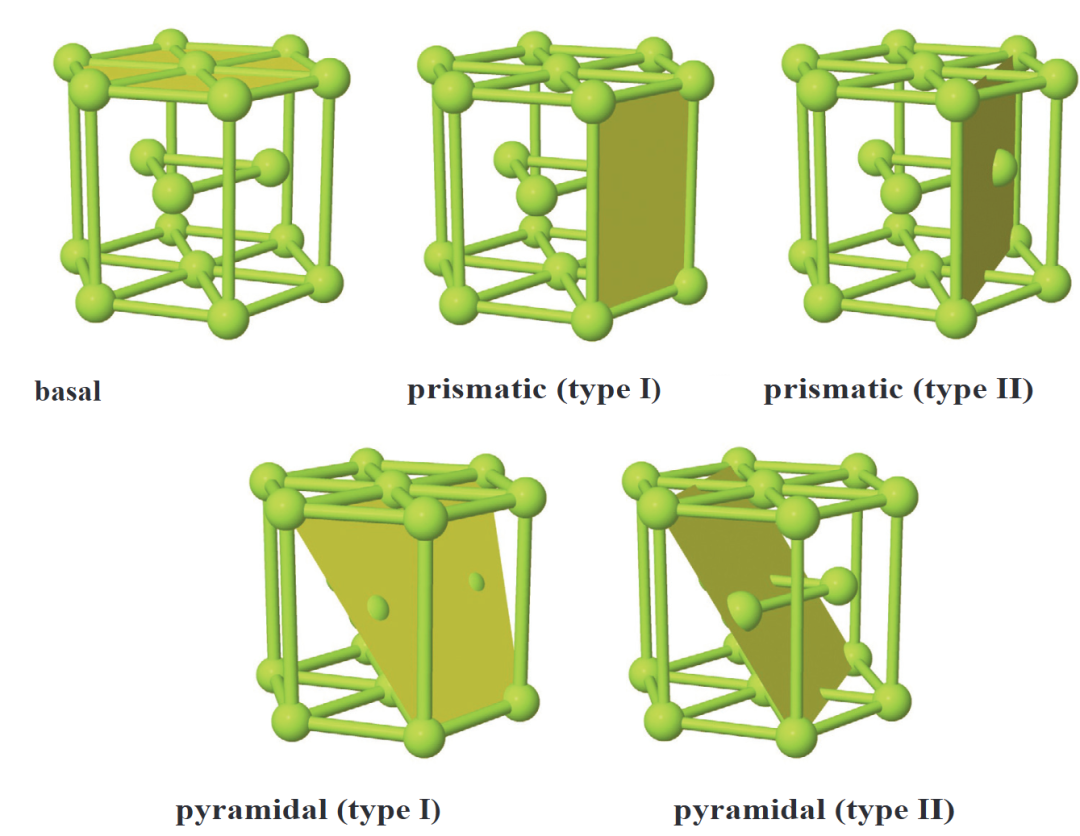
按几何结构,表面可分为纯净晶面类型(如通过构建hcp晶体基面、棱柱面等不同晶面的Slab模型,计算表面能与功函数差异)、缺陷表面(在Slab模型中引入空位、台阶等缺陷,分析缺陷形成能及对吸附的影响,如划痕、孔洞的边缘效应)和功能化表面(通过嫁接-SH、-NH₂等分子修饰表面,研究修饰后的电荷转移与结合能,如碳纳米管的共价 / 非共价修饰)。
按功能特性,可分为催化活性表面(利用DFT计算吸附能与反应能垒预测活性位点)、润湿性可控表面(通过修饰分子构型优化模拟接触角变化)和离子吸附表面(分析矿物表面与铀酰离子等的配位结构)。
其中,DFT在表面研究中扮演核心角色,可通过表面能计算确定最稳定构型,通过势能面扫描(PES)预测能量最低吸附位点,通过态密度(DOS)分析表面态对催化活性的影响,为解码表面结构与功能的内在关联提供关键工具。
DOI:10.5445/KSP/1000044741
DFT研究表面
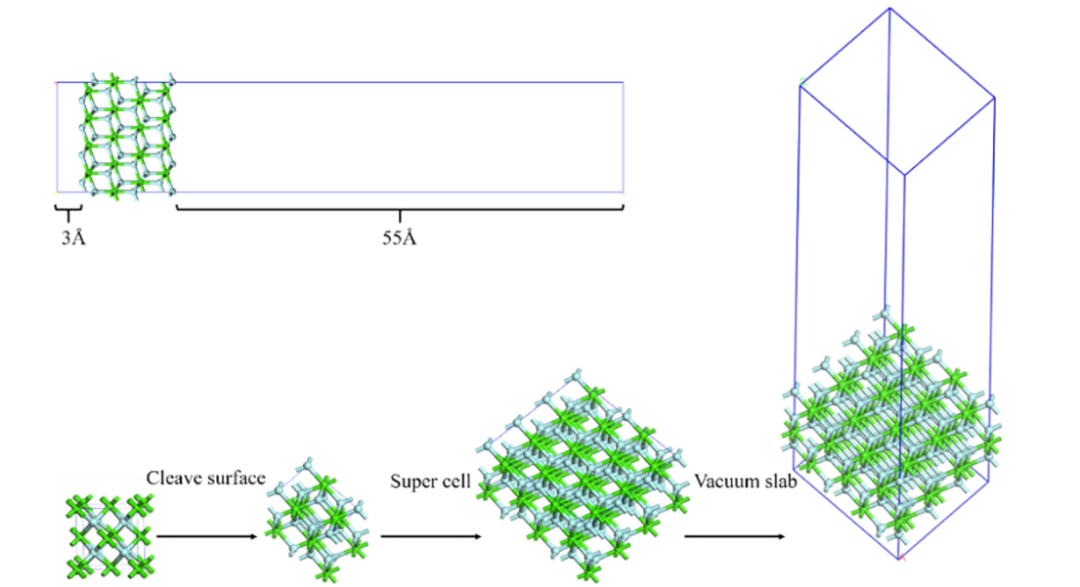
在DFT研究表面的过程中,Slab模型的构建如同为材料表面搭建 “微观舞台”:先切开晶格获得目标表面,再通过扩胞和添加≥15Å的真空层,巧妙模拟半无限体相与真空 / 溶液的界面环境,消除周期性镜像干扰,为表面科学研究搭建了精准的 “数字实验室”。
吸附构型优化则像一场分子姿态的 “选美大赛”,以卟啉在Au (111)表面为例,通过灰色球棒模型的分子与黄色金原子的键长键角优化,从平面、倾斜等五种构象中锁定最稳定的吸附姿态,直观展现分子与表面的 “互动舞步”。
而吸附能 – 结构描述符关联图堪称催化剂设计的 “导航地图”,Ni-S相的DFT计算散点与理论拟合虚线的交汇,证实Ni/S配位数可精准预测OH吸附自由能,让科研人员能透过结构参数预见催化活性,为高效催化剂设计提供量化指导。
DOI:10.3390/min13081005
经典案例:DFT研究金属表面
在DFT研究金属表面的经典案例中,科研人员以过渡金属掺杂Cu (111)催化乙酸加氢制乙醇为对象,探究Ni、Pd、Pt等元素对反应路径的调控机制。
研究首先构建Cu (111)的Slab模型,用掺杂原子替换表层Cu原子,如同为催化剂表面布置不同的 “原子岗哨”,随后通过吸附位点扫描计算乙酸分子在掺杂表面的吸附能,精准定位最易发生反应的活性点位。
接着利用NEB方法进行过渡态搜索,量化揭示CH₃CO转化为CH₃CH₂OH的能垒变化,而电子结构分析则通过d带中心位置的偏移,从电子耦合角度解释了Pd掺杂为何能降低反应能垒。
关键结论表明,Pd掺杂使速率控制步骤的能垒降低0.8 eV,其d带中心上移增强了表面与反应中间体的电子交互作用。
这一案例生动展现了DFT如何像 “分子级别的反应模拟器”,通过精准的理论计算替代传统实验试错,为高性能催化剂的设计提供从原子结构到反应能垒的全链条量化指导,让科研人员在原子尺度上解码催化过程的内在规律。

DOI:10.1039/c6ra26373a
总结
DFT在表面研究中展现出三大核心优势:一是具备原子级精度,能解析表面重构、吸附构型等实验难捕捉的微观细节;二是可高通量筛选不同表面修饰或掺杂方案对性能的影响,大幅提升研究效率;三是能通过电荷密度差(Δρ)和态密度(DOS)解码电子转移机制。
面对研究挑战,学界已发展出改进方法:如DFT+U方法修正CO在Cu表面吸附位点的误判,RPBE泛函较PW91更贴近CO吸附能实验值。
这些进展让DFT成为拆解材料表面 “关键战场” 的核心工具,为能源催化、电子器件等领域的设计提供坚实理论支撑,让科研人员在原子尺度上洞悉表面科学的本质规律。