

使用VASP软件计算材料的能带结构是第一性原理计算中的一项重要任务,广泛应用于材料科学、凝聚态物理和电子器件设计等领域。能带结构描述了材料中电子在不同k点(波矢量)下的能量分布,是理解材料导电性、光学性质和磁性等物理性质的关键。以下将详细介绍如何使用VASP计算能带结构,包括输入文件准备、计算步骤、数据处理和结果可视化,并结合多张图片进行说明。



一、输入文件准备
在进行能带计算之前,必须准备好一系列输入文件,这些文件定义了计算的参数和结构。主要的输入文件包括:
1.POSCAR:定义材料的晶体结构,包括晶格常数、原子种类、位置和基矢量。
2.POTCAR:包含赝势信息,用于描述原子的电子结构。
3.INCAR:控制计算的参数,如泛函选择、收敛标准、k点设置等。
4.KPOINTS:定义k点网格,用于能带计算。
5.CHGCAR 和WAVECAR:来自静态自洽计算(SCF)的电荷密度和波函数文件,用于非自洽计算。
1.1 结构优化(Relaxation)
在进行能带计算之前,通常需要先进行结构优化,以确保原子位置和晶格参数处于能量最低状态。结构优化的输入文件包括:
lPOSCAR:定义初始结构。
lINCAR:设置 IBRION=2 进行离子弛豫,ISIF=3 用于晶格弛豫。
lKPOINTS:设置为 0 0 0,表示不使用k点,仅进行离子弛豫。
lPOTCAR:确保赝势正确。
结构优化完成后,会生成 CONTCAR 文件,其中包含优化后的结构信息。
1.2 静态自洽计算(SCF)
在结构优化完成后,进行静态自洽计算(SCF),以获得收敛的电荷密度和波函数。SCF计算的输入文件包括:
lPOSCAR:使用 CONTCAR 文件。
lINCAR:设置 ICHARG=11,表示读取自洽迭代的电荷密度;LCHARG=.F. 和 LWAVE=.F. 表示不保存电荷密度和波函数;LORBIT=11 表示写入投影电荷密度(PROCAR)。
lKPOINTS:设置为 0 0 0,表示不使用k点,仅进行自洽计算。
SCF计算完成后,会生成 CHGCAR、WAVECAR 和 OUTCAR 文件。OUTCAR 文件中可以找到费米能级(Fermi level)和倒格子矢量(reciprocal lattice vectors)。
1.3 能带计算(Bands)
在SCF计算完成后,进行能带计算(Bands),以获得不同k点下的能量分布。能带计算的输入文件包括:
lPOSCAR:使用 CONTCAR 文件。
lINCAR:设置 ISTART=1,表示读取之前的电荷密度和波函数;ICHARG=11,表示读取自洽迭代的电荷密度;ISMEAR=0,表示无高斯函数拖尾效应;NBANDS=…,根据需要设置能带数。
lKPOINTS:根据晶体结构的空间群编号,在https://www.cryst.ehu.es/rep/repres.html 网站选择高对称点路径,并设置k点密度。例如,对于Si晶体,可以设置沿高对称线(Γ-X-W-K)的50个k点。
能带计算完成后,会生成 EIGENVAL 文件,其中存储了每个k点对应的不同能级的能量绝对数值。此外,BANDS 文件(如 bnd.dat 和 highk.dat)可以用于后续的图形化展示。
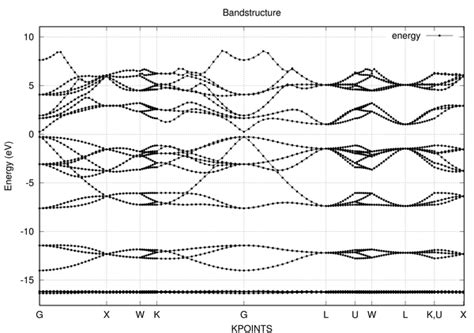
二、数据处理与结果可视化
2.1 提取能带数据
从 EIGENVAL 文件中提取能带数据,可以使用脚本工具(如 bands_plot.py 或 vaspkit)将数据转换为 bnd.dat 文件。bnd.dat 文件包含每个k点对应的能量值,可以用于Origin或其他绘图软件进行可视化。
2.2 绘制能带图
使用 vaspview 软件可以直接生成能带图,操作简单。此外,也可以使用 Origin 或 matplotlib 等软件手动绘制能带图。在绘制能带图时,需要注意以下几点:
k点路径:选择合适的高对称点路径(如Γ-X-W-K),确保能带图的准确性。
能量单位:通常以电子伏特(eV)为单位,横轴为k点路径,纵轴为能量。
能带颜色:不同颜色代表不同的能带分支,通常价带为红色,导带为蓝色。
费米能级:在图中标注费米能级(Fermi level),通常位于图中某个位置。
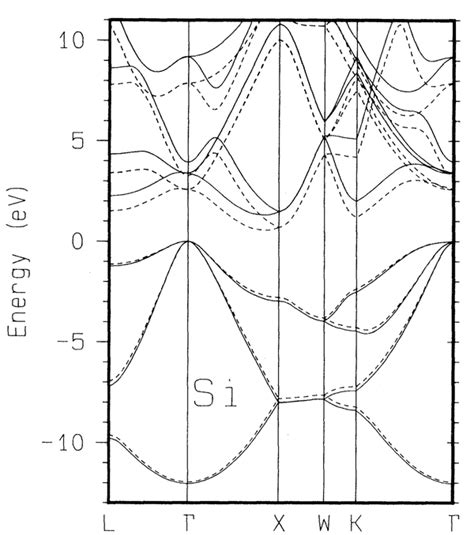
三、参数设置与注意事项
在进行能带计算时,需要注意以下参数设置和注意事项:
3.1 INCAR文件设置
ICHARG=11:读取自洽迭代的电荷密度。
ISMEAR=0:无高斯函数拖尾效应。
SIGMA=0.01:设置高斯函数的宽度,通常用于金属材料。
PREC=Normal 或Accurate:控制计算精度。
ENCUT=250 eV:设置平面波截断能,确保电子结构的准确性。
LCHARG=.F. 和LWAVE=.F.:不保存电荷密度和波函数,以节省磁盘空间。
LORBIT=11:写入投影电荷密度(PROCAR),用于能带结构分析。
3.2 KPOINTS文件设置
lKPOINTS 文件用于定义k点网格,通常使用 Monkhorst-Pack 方法生成高对称点路径。例如,对于Si晶体,可以设置沿高对称线(Γ-X-W-K)的50个k点,高对称点之间撒点数越高,能带计算越精细但计算量也越大。
3.3 收敛标准
EDIFF=1e-6:设置总能量收敛标准。
ALGO=Normal:使用标准自洽算法。
NSW=0:不进行离子弛豫,仅进行自洽计算。
四、能带结构分析
能带结构分析是理解材料电子性质的重要手段。通过分析能带结构,可以判断材料的导电性、带隙、电子结构等性质。例如:
导带底(CBM)和价带顶(VBM) :能带结构中导带底和价带顶之间的能量差即为带隙(Band Gap),是判断材料是否为半导体或绝缘体的重要依据。
能带交叉点:能带交叉点表示电子可以在不同能带之间跃迁,是理解电子结构的重要特征。