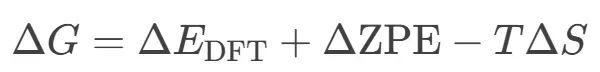压力是影响材料结构和催化性能的重要因素,可分为静水压力与气相压力。静水压力通过晶格压缩和晶相转变影响催化剂的电子结构与表面活性位,从而改变催化性能。气相压力则通过调控催化剂表面吸附覆盖度和电子结构,导致反应路径和催化机制的转变。
理论计算(如密度泛函理论DFT)通过变胞优化模拟静水压力效应,以及通过引入化学势与统计热力学方法来准确模拟气相压力的作用。
近年来的研究通过计算结合原位实验,在CO2加氢反应、甲烷水蒸气重整和CO氧化等反应中揭示了压力对催化剂结构和性能的具体影响,强调了准确考虑压力条件对于实际催化剂设计和理论与实验深入合作的重要性。
在催化研究中,反应条件(尤其是温度和压力)对催化剂的结构和性能有显著影响。压力可分为静水压(对固体材料施加的均匀机械压力)和气相压力(反应气体的分压)。
从实验和理论角度来看,二者都会引起材料结构及催化行为的变化,包括晶体结构转变、表面物种覆盖度变化以及由此导致的催化活性和反应路径的改变。
例如,将催化剂置于高压环境(如几百MPa甚至GPa的压强)下,其晶格会被压缩,从而导致晶体结构和键长发生变化。
这种结构变化可能会影响催化性能:一方面,晶格压缩会改变材料的电子结构(如d带中心位置),导致吸附能的变化;另一方面,极高的静压可能诱发晶相转变,形成不同结构的相(例如某些金属或氧化物在高压下转变为更致密的结构)。
新相的表面性质与原相不同,因而催化活性也可能提高或降低。这类因静压引起的结构转变在常规催化条件(通常压力
例如,有研究通过高压控制金属扩散来制备高密度的单原子催化剂,发现降低环境压力可以抑制金属聚集,从而稳定单原子分散。
一般而言,机械压力可以通过改变活性位的配位环境来影响催化剂性能:适度的压缩可能提升某些反应步骤的能垒或改变选择性,而过高的压力可能导致结构塌陷或失活。因此,理解静压对催化材料结构的影响对于设计稳定高效的催化剂也很重
气相压力(即反应物或产物的分压)对异相催化反应影响深远。这种压力直接决定了催化剂表面吸附物的覆盖度以及表面化学态。
“压力鸿沟”(pressure gap)是表面科学领域长期关注的问题:在超高真空(UHV)条件下研究的单晶表面与实际高压反应条件下催化剂表现往往不同。
高压环境下,丰富的气体分子使表面吸附种之间发生强相互作用,从而可能出现截然不同的机理。随着反应物或产物的压力升高,催化剂表面的吸附覆盖度增加,表面金属原子的电子结构被改变,甚至一些金属-金属键被吸附种打断,从而引发表面重构。
例如,在较高氧分压下,金属表面可能生成表面氧化物层;高氢分压下,一些金属会吸收氢形成金属氢化物。这些在高压下生成的表面物种或相会显著影响催化性能:有时表面氧化层提高反应速率,有时则抑制主反应(或引发副反应)。
又如,一氧化碳(CO)在催化剂表面的高覆盖会阻挡活性位点导致失活,但在某些情况下CO本身亦可稳定特殊的活性结构。因此,相较于真空/低压条件,高气相压力下吸附行为和反应路径都可能发生转变,催化活性和选择性也随之提升或抑制。
这在很多反应中得到验证:例如CO氧化反应中,在大气压条件下观察到与UHV截然不同的反应级数和中间体,就是因为高压下表面覆盖的O和CO使机制发生改变。
总的来说,实验研究表明确实反应环境对催化剂结构和性能有巨大的影响,需要充分考虑压力因素。
为了在理论计算中再现压力对催化剂的影响,需要分别考虑如何在模型中施加静水压以及如何引入气相的压力/分压。第一性原理计算(如DFT)提供了一系列工具来模拟这两种压力情形,使理论结果更接近实际反应条件。
在密度泛函理论(DFT)计算中,可以通过引入外部应力或改变计算盒的体积来模拟静水压力对固体的作用。具体来说,大多数DFT软件都允许对晶胞进行变胞优化:不仅原子位置优化,还让晶格参数调整,以在给定压力下找到体系的平衡结构。
这实际上是最小化焓函数H=E+PV的过程,其中E是体系的DFT总能量,PV项代表外部压强P对体积V所做的功。当对体系施加一个正的外压P并进行变胞结构优化时,模拟的结果相当于材料在该压力下的平衡结构。
如某些代码实现了“恒压计算”,直接在优化目标函数中加入PV项,从而得到在压力P下稳定的晶格常数和原子构型。如果缺乏直接的恒压功能,也可以通过计算一系列不同体积下的能量(E-V曲线),拟合状态方程(如Birch-Murnaghan方程)来估计在特定压力下的平衡体积。
但应注意,直接施加压力优化往往比事后拟合更准确。通过上述方法,理论研究者可以预测高压下材料是否会发生相变,以及晶格压缩对键长、能带结构的影响。
这些信息有助于评估静压如何改变催化相关的性质,例如活性位的键合情况或表面应力。需要指出,由于催化剂实际运行时施加纯静水压的情况少见,上述模拟更多用于探究材料在极端条件下的稳定性和结构演变,以及理解应力对催化活性的影响。
模拟气相压力主要涉及将气体分子的化学势引入计算,从而评价不同压力、温度下吸附和反应的自由能变化。DFT本征计算通常给出真空条件下的总能量,而实际反应在有限温度和压力下进行,需要考虑熵和温度-压力的影响。
一种常用方法是从头算热力学 (ab initio thermodynamics, AITD) 分析:即利用DFT计算的能量结合统计热力学,将温度T和分压P引入Gibbs自由能的计算中。在这一框架下,气相分子的自由能随压力的增加而降低,其化学势可表示为:
实际操作中,可以通过计算各种吸附态的自由能来比较其稳定性,并据此绘制
表面相图或Pourbaix图(若考虑电极电位和pH)等。
例如,对于金属表面的氧覆盖,DFT可以计算不同覆盖度O吸附层的形成能,结合O2气体在不同P,T下的化学势,得到在给定条件下哪种表面结构(洁净金属、亚稳氧覆盖还是表面氧化物)最稳定。
这种方法在文献中广泛应用:如Reuter等人早期利用此方法预测了RuO2表面在不同氧分压下的稳定相;又如近期研究对CeO2负载的Pd团簇进行了热力学分析,确定在一定温度和氧气压力下Pd氧化物物种的稳定性。
除了平衡结构,自由能计算也应用于反应路径分析:通过计算各反应步骤的Gibbs自由能变化(其中气体分子的熵项∆G会包含对数P项的贡献)
可以预测在指定温度、压力下反应的顺逆倾向以及优势路径。尤其对于吸附-脱附平衡和覆盖度相关的机理,气相压力决定了表面吸附的饱和程度,需用化学势修正DFT能量来评估真实情况。另外,在动力学模拟中,压力通过影响反应物在表面的分布而进入微观动力学模型(如微观动力学模拟中,速率常数会包含分压项)。总之,通过在理论计算中纳入气相分子的化学势和相应的熵效应,我们能够更准确地再现实验条件下的催化行为,桥接传统DFT计算与实际高压催化环境之间的差距。DOI:
10.1021/jacsau.1c00355
在CO2加氢制醇类燃料的研究中,反应气氛的压力被发现能够显著改变产物的分布。实验上观察到,在Pd基催化剂上CO2加氢往往选择性生成甲酸(HCOOH),而理论计算曾预测更倾向生成甲醇(CH3OH)。
Brookhaven国家实验室的Ping Liu团队对此差异进行了深入研究,他们意识到传统理论计算忽略了高压反应环境对表面状态的影响。2022年,该团队通过DFT结合动力学蒙特卡罗(KMC) 的方法,模拟了高H2分压下Pd(111)表面的反应。
计算揭示:在工业相关条件下,Pd(111)表面会被氢原子完全覆盖(一ML的H吸附),形成稳定的氢饱和表面。在这种高覆盖环境中,CO2加氢不再遵循先生成CO中间体再转化为甲醇的路径,而是经由甲酸路径选择性地产生甲酸。
具体而言,高氢覆盖提供了充足的表面氢,从而:(1)促进了CO2通过形成HOCO中间体直接加氢为甲酸的步骤;(2)表面氢还创造出局部的“氢空位”位点,利于甲酸的生成和脱附。这一结果与在Pd上观察到主要产物为甲酸的实验相符,解决了先前理论与实验的不一致。
该研究强调了“反应环境的重要性”:只有将高压下的表面覆盖情况纳入模型,才能正确预测催化反应的选择性。这表明在理论设计催化剂时,必须考虑实际压力下表面可能呈现的富氢或富其它吸附种的状态,否则会对主导反应路径的判断产生偏差。
压力不仅来自于反应物,同样也来自产物分子的累积。Tang等人在《Nature Catalysis》发表的研究(2022年)展示了产物气体压力如何驱动催化剂在反应过程中发生动态重构。他们研究的体系是CeO2负载的Rh催化剂在甲烷水蒸气重整(产氢)反应条件下的行为。
实验通过原位/操作X射线吸收和红外等技术观察到:最初以单原子形式分散的Rh,在H2还原气氛下聚集成纳米颗粒,但在随后加入CH4和H2O进行重整反应时,体系中生成的CO(一种反应产物)会使Rh粒子重新分散为含CO配位的低核度簇(如Rh3(CO)3)。
这一可逆转变表明CO在一定压力下对Rh物种具有强烈的稳定与剪裁作用。为理解其热力学驱动,作者结合DFT计算对不同Rh物种在各种CO分压下的稳定性进行了评估。第一性原理热力学分析显示,在500℃反应温度下,当CO分压达到约0.045 bar时,小簇状的Rhm(CO)n(m=1–3)物种的自由能低于较大的Rh纳米颗粒,自发倾向于形成碳基配位的簇。
换言之,一定分压的CO可以作为配体稳定住单原子或少原子团簇,阻止它们长大聚集。这一预测与原位光谱观测吻合:当CO产物压力升高时,Rh由无配位的金属态转变成了以CO配位的小簇,降低CO压力(例如移除CO或停止反应)又会使这些簇聚回较大的金属团。
该研究揭示了产物分压可作为“自调控”因子,动态改变活性位的结构:高CO压力下形成的Rh-CO簇虽然体积更小,但被CO饱和配位,表现出不同的催化特性和稳定性。
这强调了在计算研究催化机理时,需要考虑产物的存在及其可能施加的“反压”效应,否则可能忽略实际工作状态下催化剂逐渐演化出的真实活性相。
DOI:1
0.1038/s41929-022-00741-2
传统表面科学在UHV条件下研究CO氧化机理时,常假设金属表面保持裸露的金属态。然而,在实际CO氧化反应中,高氧分压会诱导金属表面形成氧化物,相应地改变催化性能。一项经典研究是2005年发表在《Phys. Rev. Lett.》上的工作:作者在原位表面X射线衍射实验中直接观察了Pt(110)单晶在CO氧化条件下(O2和CO的总压最高达0.5bar,温度最高625K)的表面结构变化。结果发现,根据O2与CO分压比的不同,Pt(110)表面在反应过程中出现了三种明确的结构区域:(i) 裸的金属表面 (Pt(110)-(1×1)),(ii) 薄层的同步(共格)表面氧化物,及 (iii) 薄层的不同步(非共格)氧化物。尤其是第二种共格氧化物相,仅在同时存在O2和CO且足够高的氧分压条件下才出现,是高压反应环境下稳定的表面相。DFT计算进一步指明,该表面氧化物中存在碳酸根(CO3)物种稳定其结构,也就是说只有在CO和O2共同作用下(CO氧化反应中间态)才能形成该结构。功能上,这两种在高压下形成的表面氧化物相的催化活性远高于裸露金属表面:实验测得在出现表面氧化层时CO转化率显著提升。这一发现说明,在实际一氧化碳氧化反应中,活性相很可能是被氧部分氧化了的铂表面,而非UHV研究假定的清洁铂。这从理论和实验两方面桥接了压强鸿沟:只有考虑接近大气压的氧气环境以及由此产生的表面氧化物,相才能解释工业条件下Pt催化剂的高活性。类似的高压下表面相变现象也见于Ru、Pd等金属的CO氧化反应研究。因此,在理论计算中通过含氧化物表面的模型并结合氧气的化学势,可以深入揭示高压条件下实际的催化活性相和机理,与实验观察达到一致。DOI:1
0.1038/s41467-025-56576-5
压力作为催化反应的重要条件,对催化剂结构和性能具有多方面的影响。
静水压可引起催化材料的晶格和相变,从而调控活性位的电子和几何结构;而气相分压则通过改变表面吸附覆盖度、稳定新的表面物种来显著影响反应途径和速率。
为使理论计算具有预测真实催化行为的能力,必须在模型中正确考虑压力因素:包括利用变胞DFT计算模拟静压效应,以及通过引入化学势和统计热力学框架来处理气相压力对反应自由能的影响。
近年来的多项研究已经成功地将计算与原位实验相结合,阐明了压力改变催化剂活性相和反应机理的具体方式。
例如,高H2分压下Pd表面氢覆盖导致产物选择性改变,高CO分压下促进Rh单原子催化剂的再分散,以及高O2分压下铂表面氧化相的形成提高了CO氧化活性。
这些案例生动地证明:只有同时关注实验条件和理论模型,才能全面理解和设计催化剂。对于实验研究者而言,通俗地理解理论计算中如何考虑压力,将有助于更有效地合作研究催化反应,在未来开发出更高效、条件耐受性更好的催化体系。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!