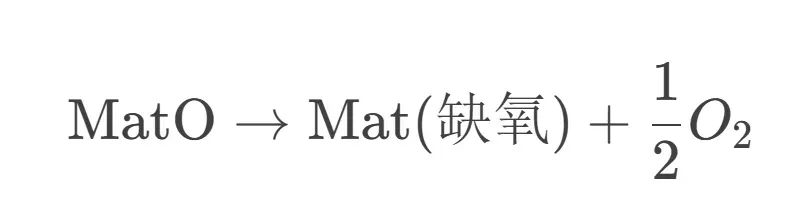材料稳定性是决定其在电池与催化等实际应用中能否长期可靠工作的关键指标,涵盖热力学、电化学及氧化等多种维度。
通过结合典型文献案例,如富锂正极材料在高电压下的氧释放行为及单原子催化剂在电化学环境下的溶解机制,展示了第一性原理计算在稳定性预测与机制揭示中的重要作用。
研究强调:在材料性能提升的同时,精准评估其稳定性并进行理论预判,是设计高效、耐久材料体系的关键步骤。
材料的稳定性是指材料在特定环境和条件下保持其化学组成和结构不发生自发改变的能力。这是材料能够应用于电池、电催化等领域的基本前提。根据作用情景和驱动因素的不同,我们可以将材料稳定性分为多个类别,主要包括热力学稳定性、电化学稳定性和氧化稳定性等。
热力学稳定性:热力学稳定性取决于材料相对于其他可能相(包括元素和其他化合物)的吉布斯自由能(或焓)。如果某材料的形成自由能较低(更负),则从热力学上更稳定,不易自发分解为其他物相。
衡量热力学稳定性常用分解能(ΔGd或ΔHd),即材料相对于在同一组成空间内所有其他可能物相的吉布斯形成能差值。当ΔGd为负时,表示该材料处于热力学稳定状态;ΔGd为正则意味着材料高于其他相的能量包络面,因而在热力学上是不稳定的,具有自发分解为更稳定相的驱动力。
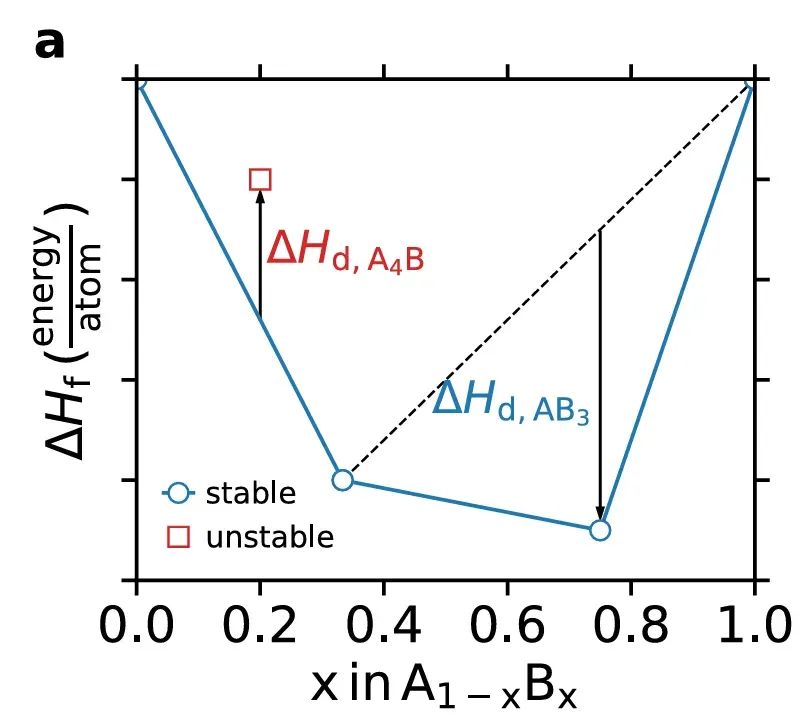
这一概念通常通过凸包构造来判断:将材料的形成焓或自由能随成分绘制,在成分-能量空间构建出下凸的能量包络面(凸包)。凸包上的化合物具有最低能量,因此是热力学稳定相,而高于凸包的化合物具有正的分解能,会倾向于分解成凸包上的组合。
如下图所示,在一个简单的二元体系中,蓝线代表由稳定化合物形成的能量凸包;位于凸包上的组分(如AB3)是稳定的,而高于凸包(如A4B)的组分将具有正的分解焓,因而热力学不稳定。
由此可见,热力学稳定性决定了材料在平衡条件下是否热力学可存活——稳定材料在没有额外驱动时不会自发转变为其它相。
DOI:10.1038/s41524-020-00362-y
电化学稳定性:电化学稳定性指材料在电化学环境(例如电池电极电位、溶液pH等)下抵抗被电氧化或电还原的能力,即材料在一定电位窗口内不发生电化学分解或相变的稳定性。
通常用电化学稳定窗口来表征:例如对于电解质,稳定窗口指其在多大的电极电位范围内不被氧化或还原。对于电极材料而言,电化学稳定性体现为材料在工作电位范围内保持其相结构不随循环电位变化而分解或发生不可逆相变。
例如,金属材料在水溶液中的电化学稳定性可用Pourbaix图(电位-pH图)来预测,在不同pH和电位下金属是以单质形式存在抑或转化为离子或氧化物。
电化学稳定性与安全和耐久性密切相关:材料在超过其电化学稳定窗口的电位下会被氧化(失去电子)或还原(获得电子)而发生腐蚀、析出或分解。
氧化稳定性:氧化稳定性是指材料抵抗与氧发生反应(氧化)的能力,通常用于描述材料在有氧环境或高氧分压、高电位等强氧化性条件下的化学稳定性。具备高氧化稳定性的材料在空气、高温或正极高电位下不易被氧气攻击或自身发生氧化降解。
例如,在电池正极中,材料需要在高充电电位(强氧化性环境)下保持晶体结构和化学状态稳定,不发生氧的释放或结构坍塌。
氧化稳定性取决于材料与氧结合的热力学驱动力:若材料在结合额外氧时能量降低,说明其易被进一步氧化(氧化稳定性差);反之则具备抗氧化的热力学倾向。氧化稳定性也与热力学稳定性相关,但特别关注与氧的反应路径和结果。
例如一些过渡金属氧化物在高电位下可能发生氧的演化(从晶格中释放O2气体)或转变为高价氧化物,体现出氧化稳定性的上限。
需要注意的是,材料的稳定性还可以有其他角度的分类(如动力学稳定性、机械稳定性等)。动力学稳定性涉及反应或相变的能垒:有些材料热力学上不稳定(能量高于凸包),但在常温下因动力学阻碍而可以长期存在(即亚稳材料)。
综上所述,材料稳定性的不同分类关注不同的失稳驱动:热力学稳定性看材料本身的能量高低,电化学稳定性涉及外加电势和环境介质(溶液)的作用,氧化稳定性则强调氧化剂(如O2或高电位)对材料的影响。理解这些稳定性类型,对于研发电池正负极材料和电催化剂材料的寿命与安全性至关重要。
随着第一性原理计算(DFT)等理论手段的发展,我们可以在原子尺度上预测并量化材料稳定性的各项指标,从而在设计新材料时预判其稳定性表现。
形成能与相图 (热力学稳定性):材料的形成能(或形成焓)是指从组成该材料的元素生成该材料的能量变化。DFT计算可以求出固体材料的形成能,用以判断其相对稳定性。
如果一个化合物的形成能在同化学体系中最低,则它将是该体系的稳定相之一。通过构建相图或凸包分析,可以确定材料是否位于能量最低的组合上。
如前述,计算多个候选物相的能量并构建凸包,判断材料的能量位置。若材料能量高于凸包(对应正的分解能),理论计算会给出它可能分解成哪些产物以及放出多少能量;若处在凸包上,则表示没有其他能量更低的组合产物,材料是全局热力学稳定的。
这种基于形成能的判据有效揭示材料热力学可行性:计算预测一个新材料若能以负形成能合成且能量低于任何可能分解产物,则它有望在实验中被制备并长期存在。
析氧/析氢电位 (OER/HER 电位):在电化学环境中,水的氧化还原反应为材料稳定性设定了基准。析氧电位(氧气析出电位)和析氢电位(氢气析出电位)通常用来表示材料相对于水的稳定性界限。
对于处于水溶液中的材料,当电极电位高于某值时,水或材料中的氧会被氧化生成O2(析氧),低于某值时水会被还原生成H2(析氢)。这些临界电位对应经典的水稳定窗口(在标准条件下pH=0时,析氢约0V vs SHE,析氧约+1.23V vs SHE),也是许多电化学过程的起点。
在理论计算中,我们可以通过比较材料含不同氧/氢含量状态的自由能差来求取析氧/析氢的临界电位。例如,对电池正极材料计算其在不同锂脱出程度下氧的释放反应:
考虑反应的反应自由能变化,将其与电子得失联系,可得到对应的氧气析出电位。如果材料在某一高电压下该反应变得自发(自由能变负),那么该电压就是材料氧失稳(氧开始析出的电位)。
实验和计算表明,很多富镍层状氧化物正极在约4.5–4.7V(对Li/Li⁺)以上会发生氧的释放。类似地,对于负极材料,在足够低的电位下(水系中约0V vs SHE),会催化水的析氢反应,从而表现出材料负极下限的不稳定性。因此,理论计算通过反应能量判定材料相对于水的氧化或还原阈值电位,帮助确定材料在电化学装置中的安全电位窗口。
能带结构与费米能级 (电子结构分析):材料的能带结构描述其价带和导带的分布,费米能级是价电子在平衡时的化学势。这些电子结构特征往往与材料的化学稳定性和反应活性相关。
首先,绝缘体与导体的电子结构差异会影响稳定性:绝缘体(存在带隙)往往化学惰性更高,因为费米能级处于带隙中,无未占据的高能电子态;而金属由于费米能级处在价带,拥有丰富的游离电子,更容易与环境发生电荷转移反应(如腐蚀)。
因此,费米能级的位置决定了材料作为还原剂或氧化剂的倾向——费米能级越高(电子化学势高)的材料,越容易失电子被氧化;费米能级低的材料则更易得电子被还原。
在腐蚀科学中,这一原理对应于金属的标准电极电势:例如锂金属的费米能级很高(对应极低的电极电势),因而在水溶液中极易氧化(与水反应放出氢气);而像金这样的贵金属费米能级较低,对应电极电势高,在环境中难以被氧化。
其次,能带结构还能揭示材料内部哪些元素或键对化学稳定性起关键作用。通过这样的能带和费米能级分析,研究人员可以判断材料在特定电子填充状态下是否易发生重键化或电荷重新分布,从而预测结构畸变或分解的可能。
例如,某些过渡金属氧化物若计算显示在费米能级处存在部分占据的d轨道与氧p轨道强烈杂化,则可能预示着易发生Jahn-Teller畸变或氧迁移,降低材料稳定性。总而言之,电子结构计算为理解材料内在稳定性提供了微观视角,揭示了不稳定迹象(如高态密度、局域未成键态),从而与化学稳定性的宏观表现相联系。
氧化分解路径 (反应机制预测):除了静态的热力学分析,理论计算还可探索材料在氧化环境中的分解反应路径。通过计算材料与氧气(或其他氧化剂)反应形成不同产物的反应能,可以预测哪些氧化产物更有可能出现,以及材料在哪些条件下会被氧化分解。
例如,对于电池正极材料,可以比较其在空气中分解为金属氧化物和氧气的多种可能路径,找出最放热(即最自发)的反应,从而确定材料的氧化分解产物和所对应的温度、电位条件。
这样的计算已经用于解释锂过渡金属氧化物在过充时的失稳:DFT研究显示,高电压下LiCoO2可能自发分解为Co3O4和O2,提高了氧化态的Co氧化物并释放氧气;类似地,富锂Mn基氧化物在深度脱锂时可能转化为尖晶石或岩盐相并伴随氧释放。这些预测与实验观察的容量衰减和气体析出相吻合。
对于催化剂材料,模拟其在工作气氛下的结构演变也十分重要。例如,在高电位酸性介质中计算过渡金属电催化剂与水/氧的反应,可以判断其是否会被氧化成高价氧化物甚至溶解成金属离子。
这样的反应路径计算能够揭示材料失效机制:是表面氧化膜的形成保护了材料(钝化),还是氧化导致材料不断溶解流失(腐蚀)。
总之,通过穷举可能的反应并比较吉布斯能变化,理论计算为材料在氧化环境中的长期稳定性提供了定量判断依据,并能指出主要的分解途径,为改进材料提供方向。
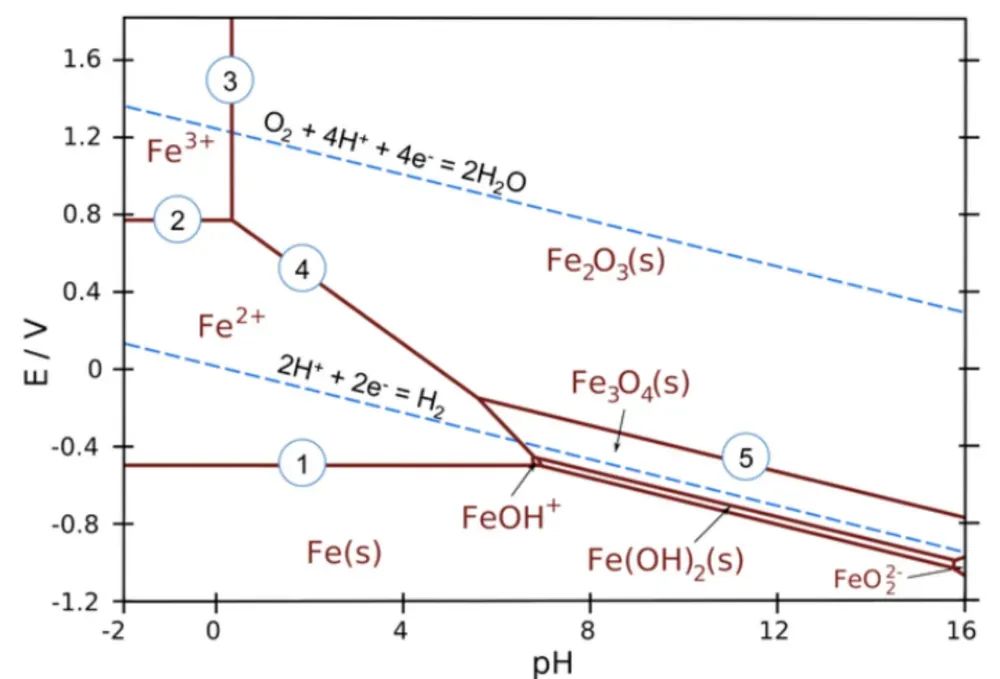
Pourbaix图 (电位-pH稳定图):Pourbaix图是一种将热力学和电化学结合的图示工具,它描绘了材料(尤其是金属及其化合物)在不同溶液pH和电极电位条件下的稳定形态。
在Pourbaix图中,纵轴为电极电位(通常相对于标准氢电极),横轴为溶液pH,根据热力学计算绘出材料处于金属单质、稳定氧化物/氢氧化物或溶解离子形式的区域边界。
这本质上是一个包含溶剂和离子物种的扩展相图,能指示材料在腐蚀环境中的平衡相。利用DFT计算材料固相的形成能,再结合水溶液中相关离子物种的已知热力学数据,可以构建理论Pourbaix图。
Pourbaix图对电催化和防腐领域尤为有用:它可直观显示某催化剂在工作条件下会保持固体形式,还是会被腐蚀溶解。
以铁-水体系为例,其Pourbaix图显示铁在中性和碱性条件高电位下转化为Fe2O3/Fe3O4等氧化物(钝化区),在中间电位和酸性条件下转化为Fe²⁺、Fe³⁺离子(腐蚀区),而在低电位下以金属态存在(免疫区),水的稳定窗口则限定了上、下电位边界。
DOI:0.1038/s41524-020-00430-3
为了更具体地说明上述概念,以下选取近期英文SCI文献中的两个代表性案例(涵盖电池和催化材料领域),介绍理论计算如何帮助判断材料的稳定性,并与实验现象相印证。
高电压锂电正极材料的氧稳定性
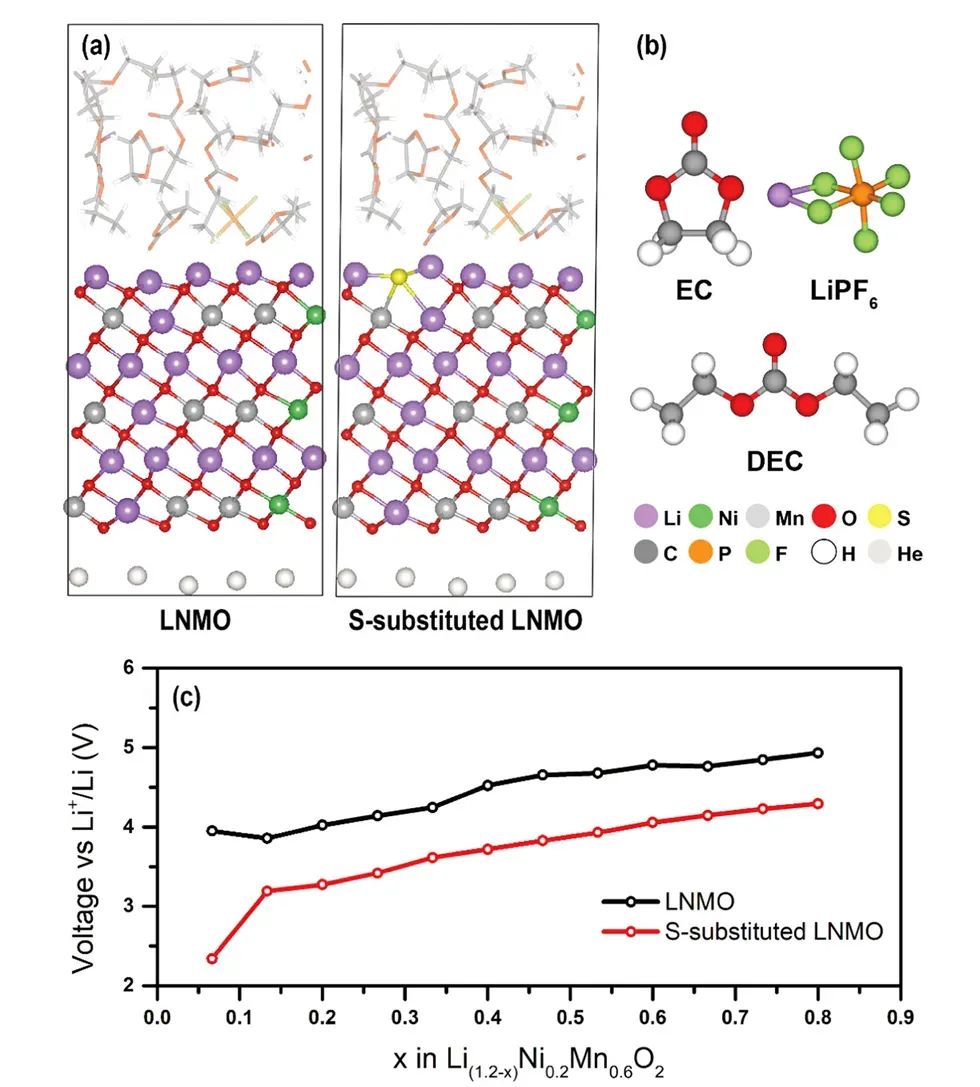
高比容量的富锂层状过渡金属氧化物(如 Li1.2Ni0.6Mn0.2O2,简称LNMO)是新一代锂离子电池正极的候选。
然而,此类材料在高充电电位下存在严重的氧气释放和结构崩解问题,表现为首周高电压滞后、容量快速衰减和电压逐步降低。
理论计算为揭示这一稳定性问题提供了关键见解:Ceder等研究者通过DFT能带结构分析发现,在富锂层状氧化物中,某些氧2p轨道沿着Li–O–Li路径形成了高能的非键合能带,直接位于费米能级附近。
这表明当提锂过多时(高电位下),氧2p电子被抽出,氧从O²⁻被过度氧化,从晶格中逃逸为O2气体。
该计算结果清晰地解释了材料热力学上为何在过高电位下不稳定:高能价带中的氧态使晶格氧缺乏束缚,易演化为气体。这一理论认识也指导了提高材料氧化稳定性的策略。
例如,有研究尝试用阴离子掺杂来降低氧的过氧化倾向。Wang等在2024年的一项工作中报道,将LNMO表层部分O²⁻用S²⁻取代后,通过DFT计算发现材料在初始脱锂阶段的平均电压从约4.0V降至3.5V。
较低的脱锂电压意味着在相同充电状态下S掺杂样品所处的氧化驱动力更小,从而推迟或减弱了氧的释放。
他们进一步的计算和分子动力学模拟也显示,S取代降低了表面氧空位的形成倾向,改善了材料结构在高电位下的稳定性。结合实验,适量S掺杂的LNMO表现出更缓慢的电压和容量衰减。
这一案例表明:理论计算不仅揭示了富锂正极材料氧化稳定性差的本质原因(氧2p非键合态导致的氧气析出),还能够通过模拟掺杂等手段预测改性方案的有效性,加速实现对高能量密度电池正极材料的稳定性优化。
DOI:1
0.1038/s41524-022-00893-6
电催化单原子催化剂的稳定性
单原子催化剂(Single-Atom Catalysts, SACs)由于最大化利用金属活性位点而在氧还原、氢演化等反应中表现出优异的活性。然而,SAC常采用过渡金属单原子锚定在载体上的结构,在强氧化性或还原性的电化学条件下,这些单原子可能发生溶出、团聚等失活,稳定性成为其实际应用的瓶颈。
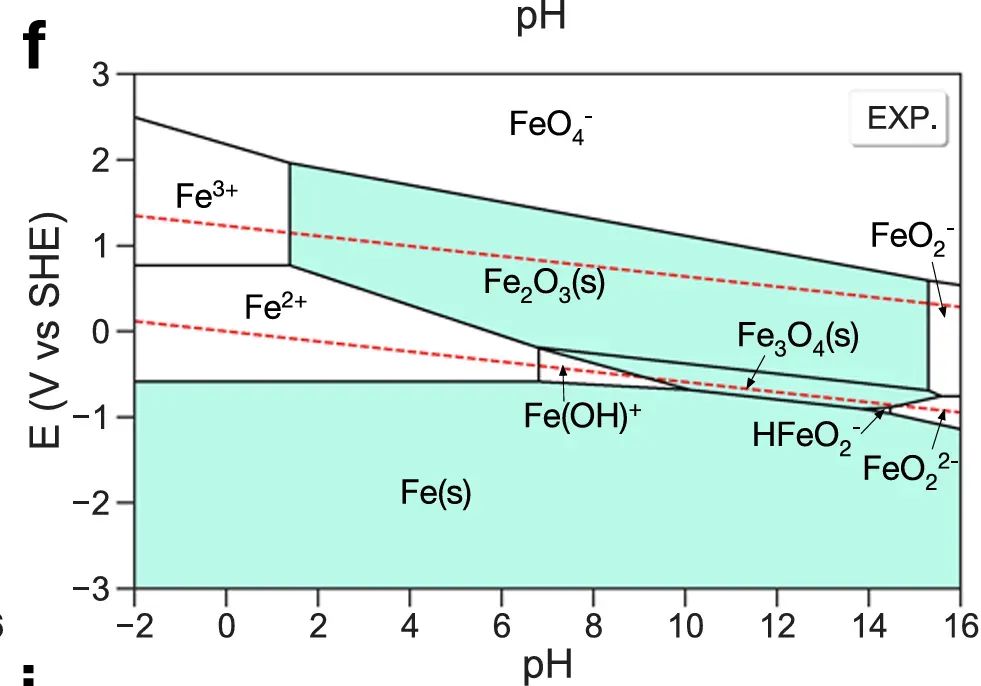
理论计算,尤其是结合热力学与电化学的Pourbaix图方法,已经成为评估SAC稳定性的重要手段。例如,Di Liberto等人在2021年的研究中采用DFT计算结合Pourbaix分析,系统预测了多种过渡金属MTN4–石墨烯单原子位点在不同pH和电位条件下的存在形态。
他们首先计算了Fe、Co、Ni等金属单原子位在被质子化、水氧化等不同表面吸附态下的自由能,进而绘制出这些SAC在电位-pH平面上的稳定区。
结果表明,在实际燃料电池阴极(低pH高电位)的工作条件下,某些候选SAC(如Fe-N-C单原子催化剂)热力学上并不稳定,金属中心将倾向于被氧化成高价阳离子并从载体上溶解。特别是在强氧化条件(高电位)下,计算预测许多潜在高活性的SAC体系会发生结构转变或金属流失。
这些预测与实验观察到的SAC活性衰减和金属流失现象相符,指出了提高SAC稳定性的必要方向,例如通过调整配位环境增加金属-载体键合强度,或在反应过程中限制金属的高价氧化。
另一个由理论证实的现象是某些材料在电催化环境下表面重构导致“原位活化”或“溶解-再沉积”等复杂过程。通过计算表面Pourbaix图,可以揭示双原子/单原子催化剂在工作电位下实际存在的表面形态与初始态可能不同。
总的来说,理论计算为SAC这类新兴催化剂的稳定性提供了宝贵的先验评价工具,能够预测单原子在何种条件下稳定存在以及失活途径,从而指导实验上改进SAC的结构设计以提升稳定性。
综上,以上案例清晰地展示了理论计算在材料稳定性研究中的关键作用:无论是电池正极材料的高压相变、固态电解质的分解限界,还是电催化剂在工作条件下的演变,计算结果均为理解和提高材料稳定性提供了定量且深入的依据。
对于实验研究人员而言,这些成果表明:借助热力学、电化学计算,我们可以在实验之前预测材料可能遇到的稳定性挑战,并据此优化材料组成和结构(如掺杂、包覆、组成调控等)来规避失稳风险。
在电池和催化领域日益追求高性能的同时,材料的稳定性要求也越来越严苛。理论计算表征手段将在这一过程中继续发挥不可或缺的作用,帮助我们设计出同时具备优异性能和长久稳定性的创新材料。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!