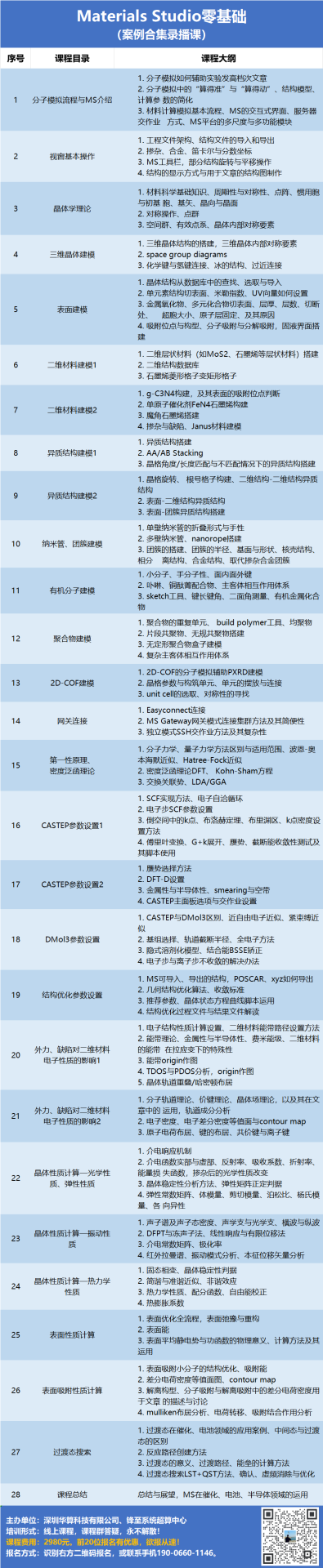
GGA+U方法是对传统密度泛函理论(DFT)的重要扩展,通过引入Hubbard U项显式处理强关联体系中局域d/f电子的库仑排斥效应,有效解决了LDA/GGA在描述过渡金属氧化物、镧系/锕系化合物等强关联材料时的系统性误差。
该方法通过修正电子自相互作用误差(SIE),显著提升了带隙预测精度、磁序描述准确性及电荷局域化表征能力。
尽管U值需经验性选取,GGA+U仍以较低计算成本实现了对材料基态电子结构、相变行为和电化学性质的可靠预测,在能源材料设计(如锂电电极电压计算)、强关联体系(如δ-Pu电子结构)和磁性材料研究中展现出不可替代的优势。
理论来源与发展背景
传统的密度泛函理论(DFT)框架,尤其是局域密度近似(LDA)和广义梯度近似(GGA),在计算材料电子结构时取得了巨大成功,但其核心近似——将多电子问题简化为单电子在有效势场中的运动——导致了对强关联电子体系的描述失效。
这类体系通常包含局域的d或f电子(如过渡金属氧化物NiO、MnO,镧系CeO₂,锕系UO₂等),其电子间存在显著的库仑排斥作用(强关联效应),而传统DFT方法无法准确处理这种相互作用。在强关联绝缘体(如NiO)中,d电子间的强库仑作用会打开带隙,但LDA/GGA因忽略动态电子关联(dynamic correlation),往往将这类材料错误预测为金属或严重低估带隙。
例如,实验测得NiO的带隙约为4.3eV,而GGA计算可能仅为0.5eV,甚至为零。这种误差源于DFT对电子自相互作用的错误描述(自相互作用误差,self-interaction error, SIE),导致电子过度离域化。强关联材料的物理性质(如磁序、轨道有序)高度依赖电子局域化。传统DFT会低估局域磁矩或错误预测基态电子构型。
例如,对于高温超导体中的铜氧化物(如La₂CuO₄),GGA可能无法重现反铁磁基态,而实验明确观察到自旋有序。此外,镧系/锕系化合物的f电子局域性在DFT中常被平滑化,导致错误的基态能量和电子分布。LDA/GGA的交换关联泛函基于均匀电子气或梯度修正模型,未显式包含电子间的短程强库仑排斥(即Hubbard U效应)。
这种作用在强关联体系中主导了电子的 Mott 绝缘体行为(如VO₂的金属–绝缘体转变)。DFT的“单粒子”框架无法描述多电子态竞争(如电荷转移、高自旋/低自旋态),从而在相图计算中产生系统性偏差。
传统DFT本质上是基态理论,难以处理激发态性质(如准粒子能隙、光谱学特征)。强关联体系中的动态屏蔽效应(dynamic screening)和多重态分裂(multiplet splitting)需要更高阶的关联方法(如GW、DMFT)才能捕捉,而LDA/GGA完全缺失这一能力。
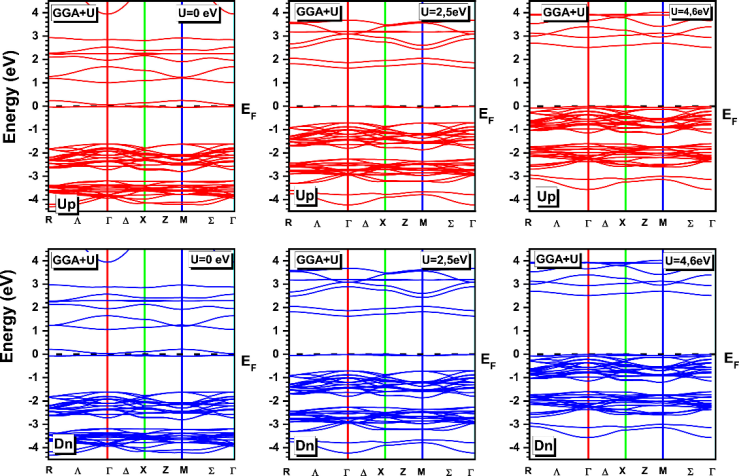

数学定义与物理意义
1、GGA+U的总能量表达式为:
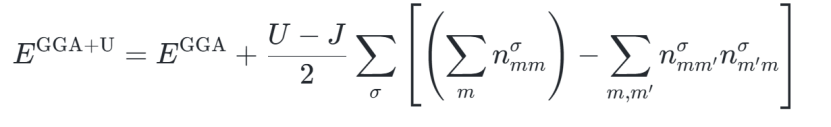
其中 为d/f轨道的占据矩阵,σ表示自旋方向。
为d/f轨道的占据矩阵,σ表示自旋方向。
2、适用体系
强关联材料(如NiO、MnO、CeO₂等过渡金属/稀土化合物)。
含局域d/f电子的体系,如高温超导体、磁性材料。
3、优势
修正带隙:相比纯GGA,能更准确地预测绝缘体的带隙。
描述电子局域化:改善电子自旋极化、磁矩和轨道有序化的描述。
计算成本较低:比杂化泛函(如HSE)或GW方法更高效。
4、局限性:
U值依赖性:U值需经验性选取,缺乏统一标准。例如,Co的3d轨道U值在不同体系中可能需调整。
仅限局域轨道:仅修正特定轨道的电子关联,对其他电子仍用GGA处理。
不适用弱关联体系:可能过度修正,导致预测偏差。
与传统GGA对比

典型应用场景
1、过渡金属氧化物
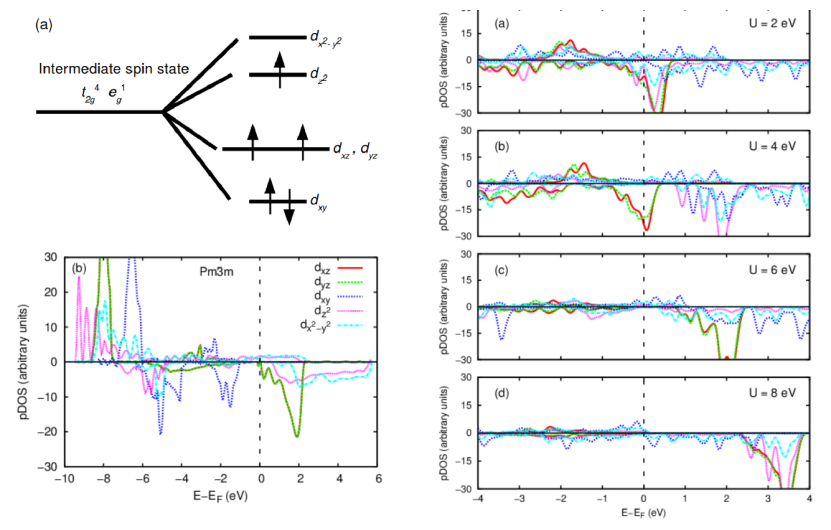
SrCoO₃中的Co³⁺离子具有强关联的3d电子,标准DFT方法(如GGA或LDA)无法准确描述其电子结构和磁性。通过GGA+U计算,作者发现SrCoO₃的基态并非传统认为的立方相(Pm-3m),而是具有Jahn-Teller畸变的四方相(P4/mbm)。
同时,研究表明,仅当U > 5 eV时,GGA+U才能正确再现Co³⁺的中间自旋态(t²g⁴eg¹),而较小的U值会导致低自旋态的误判。这一发现强调了U参数的选择对描述材料基态的关键作用,以及在晶体结构竞争中的重要性,并为后续研究提供了可靠的参数依据。
2、能源材料设计
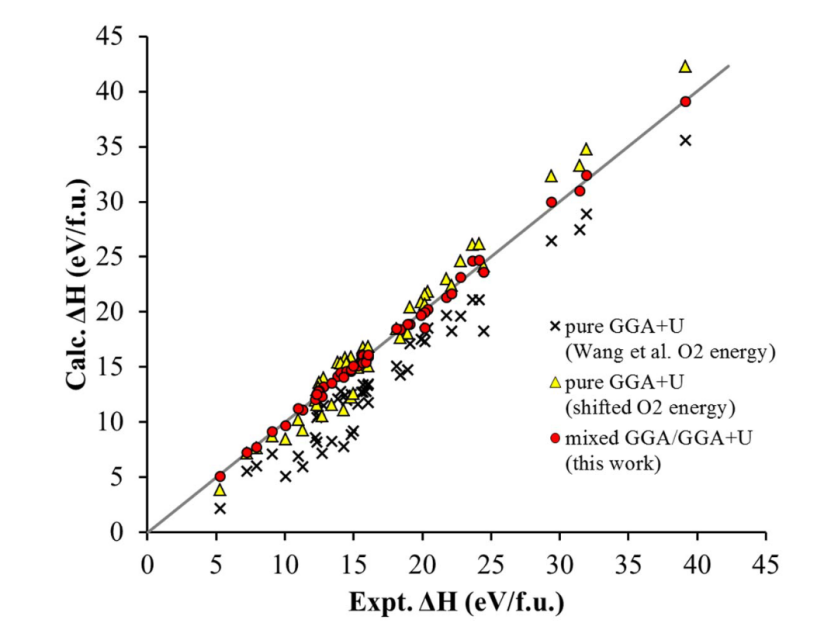
在计算转换电极电压时,混合 GGA/GGA+U 方法展现出显著优势,能大幅提升预测精度。以 LiFeF₃转换电极为例,传统 GGA 方法在预测 Li 离子嵌入电压时存在误差,这是因为其难以同时准确模拟 Fe 的离域态和 LiFeF₃的局域态 。
GGA+U 虽对过渡金属氧化物反应能量预测有一定优势,但单独使用时,计算 LiFeF₃分解反应电压的误差较大,如计算值为 3.46V,与测量电压 2.5V 相差甚远。而混合GGA/GGA+U 方法,依据不同化合物特性,用GGA计算Fe和LiF,用经调整的 GGA+U 计算 LiFeF₃,最终计算出的电压为 2.60V,与实验测量值更为接近。
与仅用 GGA+U 计算相比,混合方法将平均绝对相对误差从 7.7% 降低至 2% 以内。这表明混合 GGA/GGA+U 方法通过合理整合两种计算框架,有效减少了因电子态特性差异带来的误差,为准确预测转换电极电压提供了更可靠的途径。
3、强关联体系
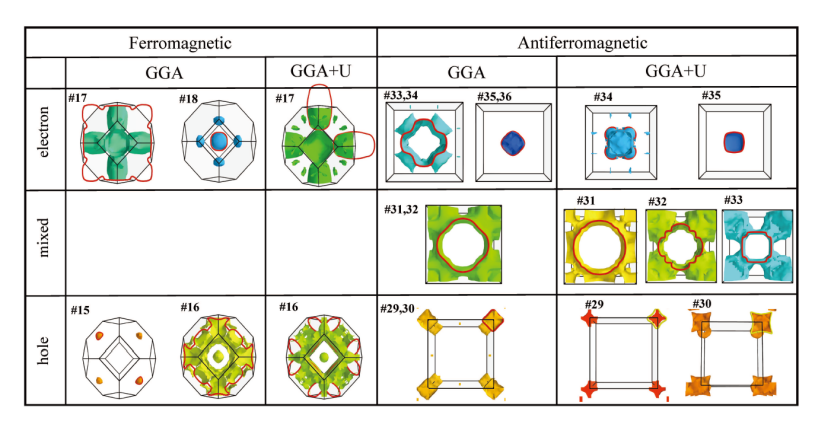
在研究δ – Pu的电子结构时,GGA+U方法在捕捉 5f 电子局域化效应方面具有重要作用。通过在 Kohn – Sham 形式体系中引入静态库仑相互作用,GGA+U 对 δ – Pu电子结构产生了显著影响。
在费米面扩张方面,相较于标准 DFT 的 GGA 计算,GGA+U 使电子和空穴费米面都出现了约 200% – 230% 的扩张。这一现象符合 Luttinger 定理,因为电子总数需保持守恒,当电子口袋体积增加时,空穴口袋体积也会相应增大以作补偿。
从电子结构角度看,添加静态库仑参数 U 后,GGA+U 使导带在费米能上方发生较大位移,上价带也发生畸变,5f 电子壳层(j = 5/2 和 j = 7/2)出现明显分裂 ,j = 5/2 电子特征主要位于费米能以下,j = 7/ 电子特征位于 4 – 6 eV 之间,这种电子分布的改变促使了费米面的扩张。
写在最后