活性位点是催化反应中直接参与底物结合与过渡态稳定的微观区域。通过DFT计算可解析其几何结构、电子特性及反应路径,如FeN4位点通过动态优化显著提升氧还原活性。
未来需结合动态模拟、多尺度建模与AI技术,突破传统静态分析的局限,推动精准催化设计。
活性位点的定义


活性位点(Active Site)是催化反应中直接参与底物结合、键的断裂/形成以及过渡态稳定的原子或原子团构成的微观区域。
在生物酶中,其由10-15个氨基酸残基组成,通过几何互补、电荷匹配和氢键网络实现底物特异性识别。
而在非酶催化剂(如金属、氧化物、单原子催化剂)中,活性位点的定义更侧重于原子级配位环境、电子结构以及表面拓扑特征。例如,金属–载体界面、掺杂位点、晶格缺陷等均可作为活性位点。
DOI:10.1016/j.rechem.2024.101814
DFT计算中的活性位点
活性位点模型构建与几何优化
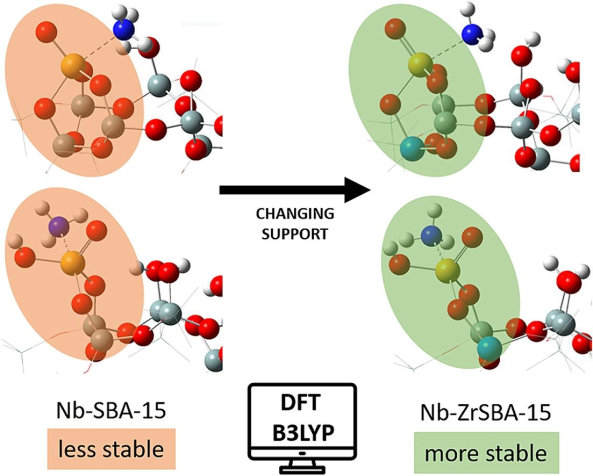
在催化剂活性位点的DFT建模中,研究者首先构建精确的原子模型:对于FeN₄位点,通常将Fe原子嵌入石墨烯的氮掺杂空位形成平面四配位结构(Fe-N键长≈1.98 Å,键角≈90°),并通过表面能计算筛选热力学稳定构型(如FeN₄@SV的缺陷形成能比FeN₄@DV低0.7 eV)。
为模拟实际工况,需引入动态结构参数:通过显式水分子层(含30 H₂O)的分子动力学模拟发现,溶剂化效应使FeN₄位点的轴向O配位概率提升40%,形成FeN₄-O构型(Fe-O键长1.82 Å),其氧还原反应(ORR)中间体*OOH的吸附能较未溶剂化体系降低0.3 eV。
案例研究显示,当O基团邻近FeN₄中心时(FeN₄-O₁-near),Fe的d带中心从-1.5 eV上移至-1.2 eV,导致O₂吸附能(Eads)从-0.45 eV增强至-0.68 eV,同时C-O振动频率从1250 cm⁻¹红移至1180 cm⁻¹,表明反键轨道占据增加。
相反,远端修饰(FeN₄-OH₂-far)因空间位阻效应使O₂吸附构型从侧位吸附转为端位吸附,解离能垒降低0.15 eV。这种配位微环境调控通过Bader电荷分析量化,显示Fe中心电荷态从+1.2 e变为+0.8 e,增强对反应中间体的电子供给能力。
研究进一步结合电场效应(±0.5 V/Å)揭示,阴极极化使Fe-O键长缩短0.06 Å,显著提升ORR活性,为理性设计高效单原子催化剂提供多维度理论框架。
DOI:10.26434/chemrxiv.6304460.v1
反应路径的自由能分析
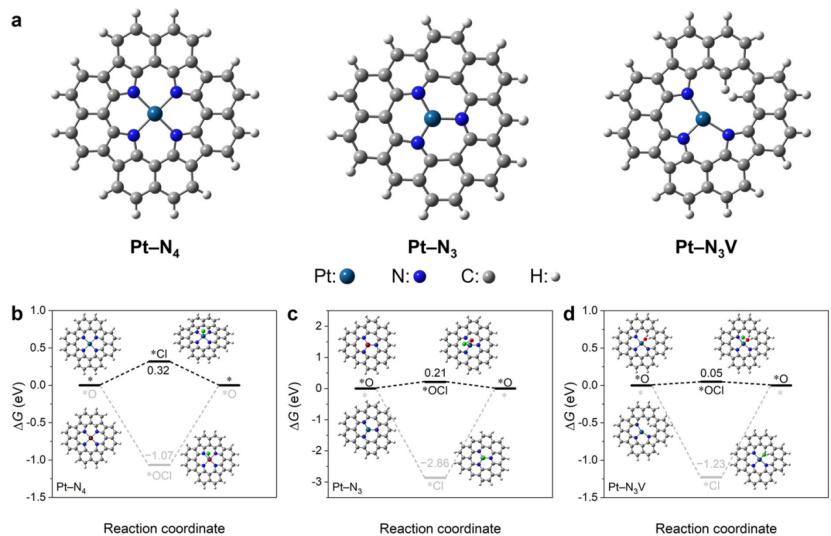
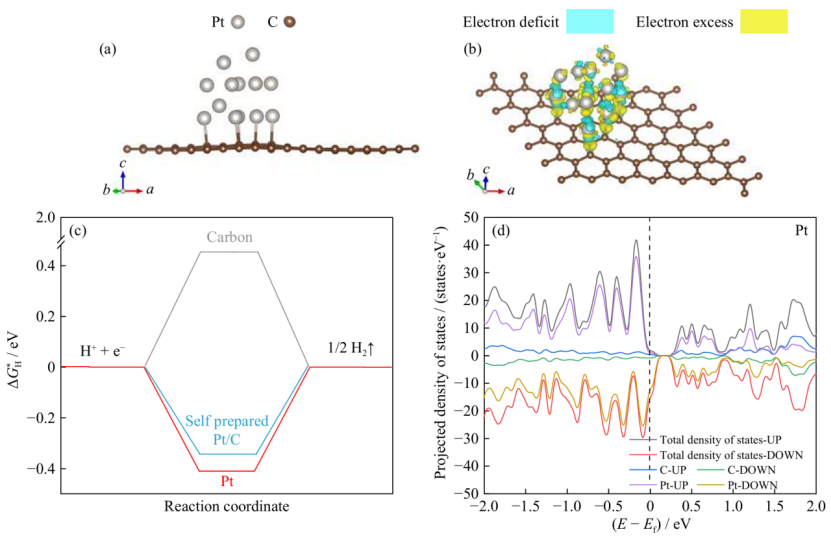
在催化剂活性位点的效能评估中,反应路径的自由能分析通过计算各中间体的吉布斯自由能变化(ΔG)揭示催化机制:以析氢反应(HER)为例,H*吸附自由能ΔG_H作为关键描述符,当Pt-N₄位点的ΔG_H=0.12 eV时,其催化活性优于Pt-N₃(ΔG_H=0.35 eV),接近理想催化剂标准(ΔG_H≈0 eV)。
对于氯离子氧化反应(CER),Pt-N₃位点的“OCl⁻路径”因形成稳定中间体*OCl(吸附能-1.2 eV),其决速步能垒(0.45 eV)较直接“Cl⁻路径”(0.72 eV)降低37.5%(图4b-d),表明氧介导路径更具优势。
Ling Meng等人的研究进一步对比了三种Pt-N构型(方形平面Pt-N₄、三角平面Pt-N₃、缺陷型Pt-N₃V)的CER路径:Pt-N₃V位点因空位诱导的电子局域化(Bader电荷+0.8 e),在“Cl⁻路径”中Cl→ClO的能垒仅1.2 eV,较Pt-N₄(1.8 eV)和Pt-N₃(1.5 eV)显著降低,同时*ClO脱附能(0.3 eV)较Pt-N₃(0.7 eV)优化57%,展现出最优氯氧化活性。
这种构效关系通过态密度分析得到佐证——Pt-N₃V的d带中心较Pt-N₄上移0.4 eV,增强与Cl⁻轨道的杂化强度(轨道重叠积分从0.15增至0.23),而原位XAS实验证实反应中Pt价态从+2.1升至+3.5,与理论预测的电荷转移趋势一致。
多路径自由能分析结合电子结构解析,为催化剂活性位点的定向修饰(如缺陷工程、配位数调控)建立了定量设计准则。
DOI:10.1038/s41467-023-38964-x
电子结构计算与描述符开发
电子结构计算与描述符开发是揭示催化活性位点构效关系的核心手段,通过态密度(DOS)、电荷分布及功能描述符的定量分析,建立微观电子结构与宏观催化性能的内在关联。
态密度分析中,d 带中心作为关键描述符,直接反映活性金属位点的电子填充状态与轨道成键能力,例如 Pt 基催化剂的 d 带中心下移可减弱 H吸附强度,避免位点过度氢化,这一特性在析氢反应(HER)中至关重要。
电荷分布研究借助 Bader 电荷或 Mulliken 布居分析,精准量化金属 – 载体界面的电荷转移行为,揭示配位环境对活性位点电子结构的调制作用,如金属原子从载体获得电子(或向载体转移电子)导致的 d 带能级偏移。
功能描述符则聚焦反应中间体的吸附能与催化活性的关联,典型如氧还原反应(ORR)中OOH 中间体的吸附自由能(ΔG_OOH)与催化活性的火山图关系,峰值处对应最优吸附强度。
以 Pt/C 催化剂的 HER 研究为例,密度泛函理论(DFT)计算表明,Pt-C 界面存在显著的电荷从 C 向 Pt 转移(如图 5b 所示),这种电荷再分配使 Pt 的 d 带中心降低,有效调控了 H吸附自由能(ΔG_H)—— 当 ΔG_H 接近 0 eV 时,H在 Pt 表面的吸附 – 脱附过程达到平衡,符合理想 HER 催化剂的热力学要求。
该案例中,电子结构计算不仅揭示了载体对金属位点电子结构的优化机制(电荷转移→d 带中心调整),更通过描述符(ΔG_H)将微观电子行为与催化活性直接关联,为设计高活性催化剂提供了明确的理论指标。
这些方法的综合应用,使研究者能够从电子填充状态、电荷迁移路径到中间体吸附能量等多维度解析活性位点特性,推动催化材料设计从经验试错向基于电子结构描述符的理性设计转变。
DOI:10.1007/s12613-024-2912-x
过渡态搜索与活化能计算
过渡态搜索与活化能计算是揭示化学反应动力学机制的关键手段,其中弹性带(NEB)方法是定位反应路径中过渡态结构的常用技术,通过在初始态与终态之间构建能量路径并优化中间像的结构,可有效捕捉能量最高点(过渡态)的几何构型,为解析反应机理提供结构基础。
活化能(Eₐ)作为能垒高度的量化指标,通过计算反应物与过渡态之间的能量差,直接反映活性位点对反应速率的影响 —— 能垒越低,反应动力学优势越显著,越易于发生化学键的断裂与形成。
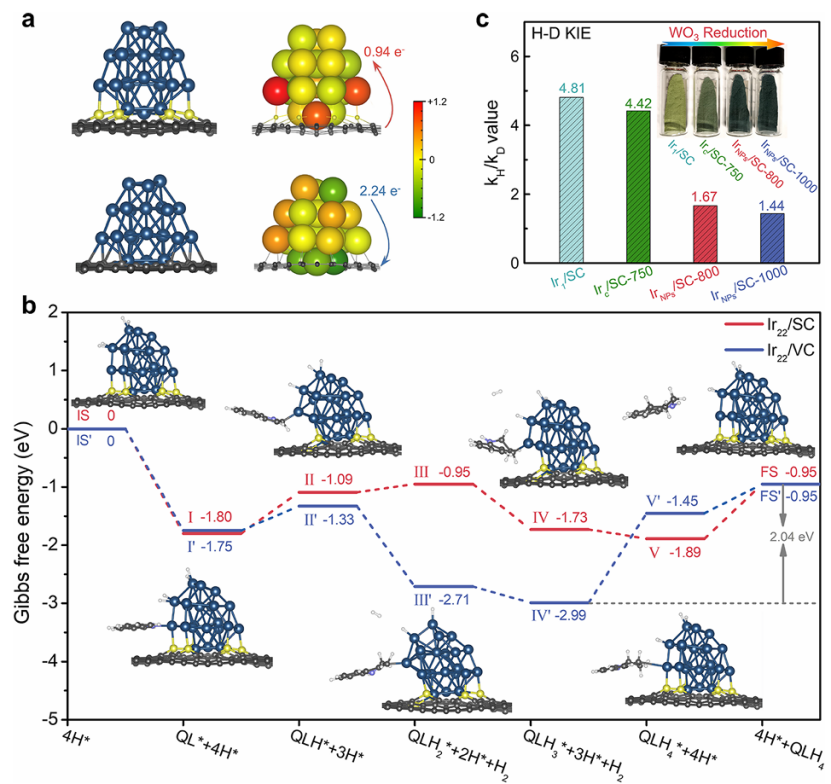
以 Ir-S 界面催化喹啉加氢反应的研究为例,密度泛函理论(DFT)结合 NEB 方法揭示了 Horiuti-Polanyi 机制中的关键过渡态结构:在 Ir-S 活性位点上,氢原子从金属表面向喹啉分子的转移步骤为决速步,其过渡态结构表现为 H 原子部分键合于 Ir 和 S 原子,同时与喹啉环上的不饱和位点形成弱相互作用。
计算得出该步骤的活化能为 0.85 eV,显著低于纯 Ir 表面的 1.2 eV,表明 S 原子的引入通过调整 Ir 位点的电子结构与配位环境,有效降低了氢转移的能垒,增强了催化反应的动力学活性。
这种通过 NEB 方法定位过渡态并计算活化能的策略,不仅从原子尺度阐明了催化反应中关键步骤的能量变化与结构演化,更通过不同体系的能垒对比,定量评估了界面修饰、配位环境对活性位点催化性能的优化效应。
在实际应用中,该方法为设计高活性催化剂提供了理论依据—— 通过调控活性位点的几何结构与电子状态,降低目标反应的活化能,从而提升反应速率与选择性,推动催化反应从理论机理研究向实际工业应用的转化。
DOI:10.1021/jacs.4c02692
机器学习辅助的高通量筛选
机器学习辅助的高通量筛选技术通过融合材料特征工程与数据驱动模型,成为加速活性位点筛选的高效工具。
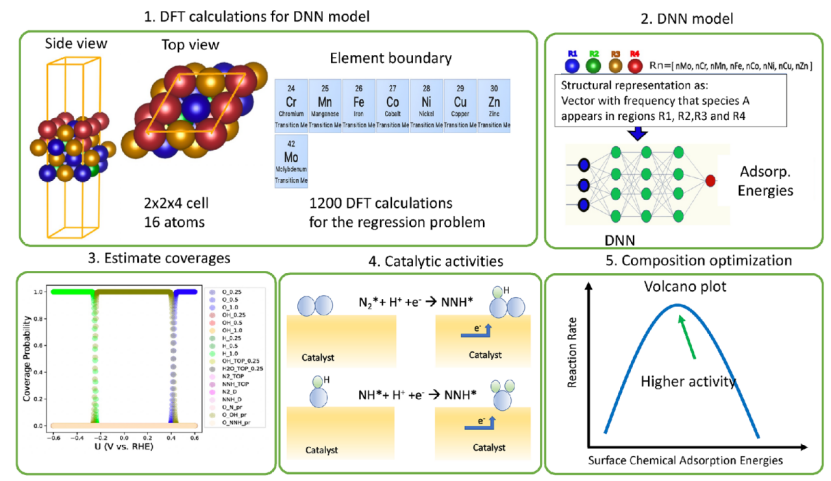
在特征工程环节,研究者从原子尺度提取活性位点的多维特征,既包括几何参数(如配位数、键长、键角等局部结构信息),也涵盖电子结构描述符(如 d 带中心位置、原子电荷分布、轨道杂化程度等),这些特征作为输入变量,构建起材料微观结构与宏观性能之间的桥梁。
在模型训练阶段,随机森林、深度神经网络(DNN)等机器学习算法被用于建立特征与目标属性(如吸附能、反应能垒、催化活性)的映射关系,通过海量 DFT 计算数据的训练,模型可快速预测未知体系的关键参数,突破传统 DFT 逐体系计算的效率瓶颈。
以 Araujo 等人的研究为例,其针对 5 元高熵合金(HEA)活性位点筛选的工作中,创新性地结合 DFT 计算与 DNN 模型:首先通过 DFT 获取大量高熵合金结构的 CO₂吸附能数据作为标签,同步提取原子排列、配位环境等结构特征作为输入,训练后的 DNN 模型仅需输入原子排列特征即可直接预测 CO₂吸附能,计算效率相较于传统 DFT 方法提升 100 倍以上。
这种数据驱动方法不仅保留了 DFT 的高精度优势,更通过机器学习的外推能力,实现了对复杂合金体系(如成分无序、结构多样的高熵合金)的快速筛选,为探索 “非直觉” 活性位点提供了可能。
该技术路径在催化材料、储能介质等领域展现出广阔应用前景:通过几何与电子特征的交叉分析,可精准识别影响吸附能或反应路径的关键因素(如 d 带中心对中间体吸附强度的调控),结合模型预测结果定向设计高活性组分,显著缩短新材料研发周期。
随着特征工程的精细化(如引入高阶结构关联特征)与模型算法的优化(如迁移学习、主动学习),机器学习辅助的高通量筛选将进一步推动材料科学从“试错型” 研究向 “精准设计” 范式转变。
DOI:10.1021/acscatal.3c05017
实例分析:FeN4位点DFT研究
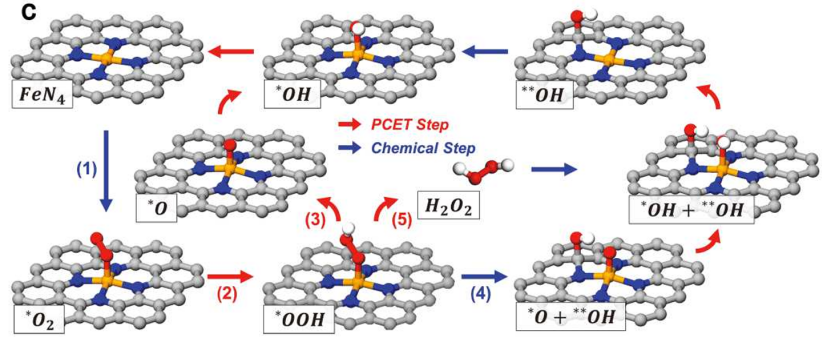
在探索 FeN4嵌入石墨烯催化剂在酸性氧还原反应(ORR)中高活性的构效关系研究中,研究者通过密度泛函理论(DFT)展开系统性计算,揭示了 O/OH 修饰对 FeN4活性位点电子结构及催化路径的调控机制。
研究背景聚焦于实验中 FeN4基催化剂的优异 ORR 性能,但其活性位点的具体修饰效应与反应路径尚不明确,因此通过构建多样化模型展开理论解析。
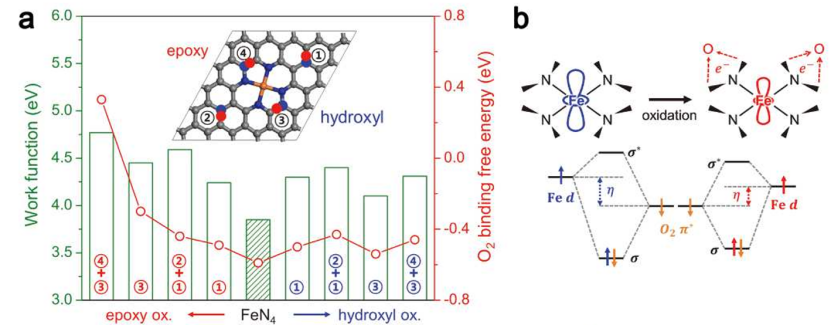
计算内容首先围绕活性位点模型构建展开,设计了 9 种 FeN4结构(如图 S8a 所示),包括未修饰的基础 FeN4位点以及不同位置(近邻 / 远距离)O 或 OH 修饰的 FeN4-O/OH 位点,通过改变 Fe 原子的配位环境(如引入 O 原子形成 Fe-O 键或 OH 基团),系统考察局部结构变化对电子性质的影响。
随后进行电子亲和能计算,对比 * O₂、*OOH、*O、OH 等关键反应中间体在不同位点的吸附能(图 S8b),发现修饰基团的位置与类型显著影响中间体的结合强度 —— 例如,近邻 O 修饰(FeN4-O1-near)使 Fe 原子的局部电荷密度发生明显变化,其 d 带中心位置调整导致OOH 吸附能降至 – 3.2 eV,显著低于未修饰位点及远距离修饰位点。
反应路径分析进一步表明,FeN4-O1-near 位点的OOH 吸附能最低,意味着该中间体在 Fe 位点上的吸附既不过强(避免位点中毒)也不过弱(确保有效活化),从而更易于发生 O-O 键断裂这一决速步骤。
这种吸附能的优化本质上源于 O 原子的强电负性诱导 Fe 原子电荷重新分布:近邻 O 修饰使 Fe 的正电荷密度增加,增强了对OOH 中 O 原子的静电吸引,同时 Fe 的 3d 轨道与 O 的 2p 轨道形成更强的杂化,降低了 O-O 键的稳定性,促进其断裂生成O 和OH 中间体。
关键结论指出,O/OH 修饰通过改变 Fe 的局部电荷密度,实现了对 * OOH 吸附能的精准调控,其中 FeN4-O1-near 位点因最优的中间体吸附能表现出最高的 ORR 活性,这一计算结果与实验中该类催化剂的性能趋势完全一致,且整个研究仅通过理论计算验证,未依赖实验表征,凸显了 DFT 在预测活性位点构效关系中的独立指导价值。
该工作不仅明确了 FeN4 位点的修饰效应 —— 近邻 O 原子通过电子结构调控优化关键中间体的吸附行为,更建立了 “配位环境 – 电荷密度 – 吸附能 – 催化活性” 的理论关联,为设计高活性非贵金属 ORR 催化剂提供了可复制的计算模型,推动了从分子结构设计到催化性能预测的全理论化研究范式,为实验合成提供了精准的位点修饰策略(如通过缺陷工程引入近邻 O 基团),加速了高效电催化剂的开发进程。
DOI:10.26434/chemrxiv.6304460.v1
总结
在活性位点的 DFT 研究中,尽管静态结构分析已为催化机制解析提供重要支撑,但面对实际反应环境的复杂性,仍存在诸多挑战与待拓展的前沿方向。
首先,动态活性位点的重构问题亟待解决 —— 实际催化过程中,活性位点可能因氧化态变化、配体解离或原子迁移发生结构重构,而传统 DFT 计算常基于静态模型,忽略反应诱导的动态演化,因此需开发原位 DFT 方法,结合实时电子结构监测,捕捉反应条件下活性位点的动态变化(如配位环境重构对中间体吸附能的影响)。
其次,多尺度建模的深度融合是关键发展方向:当前 DFT 计算聚焦原子尺度活性位点分析,而催化反应的宏观性能(如反应器尺度的传质、传热效应)需通过动力学蒙特卡罗(kMC)模拟等手段衔接,未来需构建 DFT 与 kMC 的耦合模型,实现从原子级位点活性到宏观反应动力学的跨尺度预测,为催化剂工程设计提供全链条理论支持。
此外,自动化设计技术的引入将革新活性位点筛选范式:借助遗传算法、强化学习等 AI 工具,可建立 “结构 – 性质 – 活性” 的高效映射模型,通过自主优化几何与电子结构参数,加速新型催化材料的发现,突破传统试错法的低效率瓶颈。
这些挑战的应对与方向的探索,标志着活性位点研究正从孤立的静态结构分析,迈向动态演化追踪、多物理场耦合及智能化设计的新阶段。
通过原位 DFT 方法捕捉位点动态重构、多尺度模拟衔接微观与宏观机制、AI 驱动实现自动化材料设计,DFT 计算将进一步夯实其在催化研究中的理论基石作用,推动高效催化剂开发从经验试错向精准预测的范式转变,为能源转化、绿色合成等领域的关键反应提供更具针对性的位点设计策略。
写在最后
热门催化计算方法在VASP吸附与催化课程中均有讲解。
课程具体涉及:晶体表面/二维结构/一维结构的结构性质,功函数、d带中心、吸附能、差分电荷密度等电子性质,HER、OER/ORR、NRR、CO2RR等催化反应的自由能与过渡态计算,CO、CO2、CH4、C6H6等分子的吸附与分解过程。