

说明:电负性量化原子吸引成键电子能力,通过Pauling、Mulliken等经验模型及DFT量子力学方法计算。
其周期性呈同周期递增、同族递减规律,相对论效应可使超重元素电负性异常。理论模型从键能关联发展至结合电离能、电子亲和能,现借助DFT和相对论校正提升精度,为材料设计与反应机理研究提供支撑。
什么是电负性?
电负性(Electronegativity)作为化学领域的核心概念,用于量化原子或化学物种在分子或晶体环境中吸引成键电子的能力,其本质反映了原子对电子的束缚强度与电子云的分布特征。
这一参数在预测化学键极性、评估化学反应活性以及阐释材料物理化学性质等方面具有关键作用。
尽管电负性无法通过实验直接测量,但其数值可通过Pauling标度、Mulliken模型等理论计算框架间接定义——这些模型通常基于键能数据、电离能与电子亲和能的综合考量,通过量化原子在成键过程中的电子偏移程度实现对电负性的半经验或理论模拟。
从量子化学视角看,电负性与原子的电子密度分布、轨道能级等微观参数密切相关,现代计算方法可进一步通过电子结构分析揭示电负性与物质宏观性质的内在关联,为化学键理论、材料设计及反应机理研究提供重要理论支撑。
基本定义与化学意义
通过电负性差异(Δχ)可预测键的极性——当Δχ>1.7时通常形成离子键,反之则以共价键为主;从量子化学视角看,电负性并非原子的固有常数,而是随化学环境动态变化,例如氧化态升高或配位结构改变时,原子对电子的束缚能力会相应调整,这种动态特性源于电子密度分布与轨道能级的协同变化。
在计算化学范畴中,电负性被Allen视为 “第三维元素周期表” 参数,架起了电子结构与宏观性质的理论桥梁,通过整合这些微观参数可构建描述原子成键行为的量化模型。
由于电负性无法通过实验直接观测,其数值需借助Pauling标度、Mulliken模型等理论框架,通过键能数据、轨道能量等可计算量间接推导,这种间接性本质要求理论模型在处理实际体系时,需结合电子结构计算与光谱实验数据进行交叉验证,从而精准刻画原子在不同化学环境中的电子吸引能力,为化学键理论、反应机理分析及功能材料设计提供关键理论支撑。

DOI:10.1201/9781003396512
电负性理论模型
在计算化学领域,电负性的理论模型按其构建基础可分为经验尺度与量子力学模型,不同模型通过整合可计算物理量实现对原子吸引成键电子能力的量化描述。
1932年Pauling提出的经验模型基于热化学键能数据,将电负性差与实测键能和纯共价键能的差值关联,以F元素电负性4.0为参考,直观反映键的部分离子性,适用于主族元素键极性预测,但其局限性在于忽略原子电荷变化与杂化效应,且对过渡金属体系适用性不足。
1934年Mulliken提出的绝对电负性模型以原子本征性质为基础,将电负性与价电子能量直接关联,在DFT中其负值近似于金属功函数,但依赖精确的IP和EA数据,尤其对负EA元素计算误差较大,且未处理价态依赖性问题。
1989年Allen提出的光谱电负性模型基于自由原子价电子能量,数据源自NIST原子能级数据库,该模型将电负性定义为价壳层电子的平均单电子能量,能合理解释元素周期表中金属–非金属的对角线分离现象,与Pauling尺度高度吻合,但受限于过渡金属d轨道复杂的径向分布,在该类体系中适用性有限。
1978年Parr等人将电负性纳入DFT框架,衍生出绝对硬度的概念,该模型基于严格量子力学理论,遵循电负性均衡原理,认为分子内电负性均匀分布并驱动电荷转移,为计算电荷分布提供自洽方法,但其计算成本高且结果依赖泛函选择。
不同模型的对比揭示其核心差异:Pauling模型以键能差异为基础,优势在于直观且广泛适用于主族元素,局限是经验性强且忽略杂化与电荷效应;Mulliken模型以IP和EA为基础,建立绝对尺度且理论严谨,但对数据精度要求高且价态依赖性显著;Allen模型以价电子电离能为基础,高精度匹配实验且解释周期性规律,过渡金属适用性差是主要局限;DFT模型以总能量导数为基础,普适性强且可处理复杂体系,但计算复杂度与泛函依赖性制约其广泛应用。
值得注意的是,所有模型均通过电离能、电子亲和能或轨道能量等可计算量间接定义电负性,避免直接实验测量,形成了从经验近似到量子力学第一性原理的完整理论体系,为化学键极性分析、反应活性预测及材料电子结构设计提供了多尺度理论工具。
DOI:10.1201/9781003396512
理论模拟方法
在计算化学领域,电负性的理论模拟依托量子力学第一性原理,形成了多尺度的计算框架,主要包括基于轨道能量、密度泛函理论及相对论校正的方法。
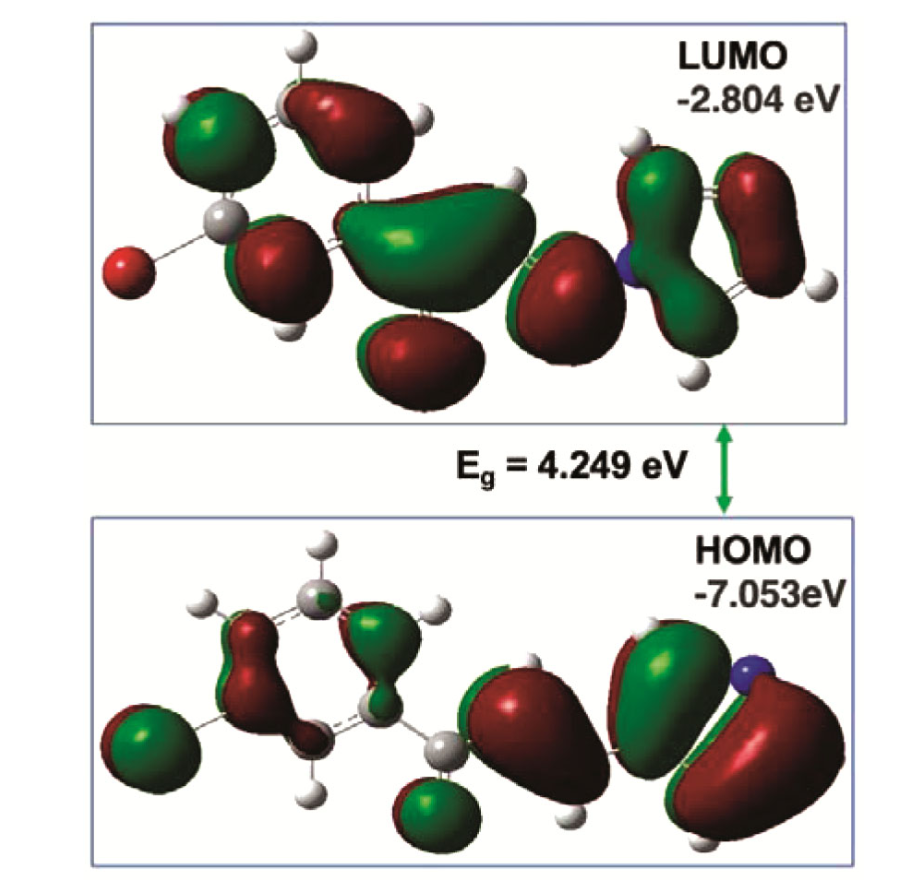
基于轨道能量的方法以Koopmans定理为核心,通过分子轨道能量间接推导电负性,其数学框架为χ=(I+A)/2,其中电离能I≈-EHOMO、电子亲和势A≈-ELUMO,EHOMO和 ELUMO分别为最高占据和最低未占分子轨道能量。
模拟流程包括通过Hartree-Fock或DFT计算分子波函数、提取轨道能量并代入公式,例如对有机分子C₉H₆O₂的计算得到χ=3.68 eV,与实验趋势一致。该方法计算具有高效性,但Koopmans定理忽略电子相关效应,需借助耦合簇等高阶方法校正以提升精度。
DFT为电负性模拟提供了自洽的理论框架,其核心是通过Kohn-Sham方程求解电子密度ρ,进而从能量泛函导出化学势μ,定义电负性χ=-μ。
在原子尺度,Perdew等采用GGA泛函计算H至Cu的电负性,误差小于0.1 eV;在分子尺度,自洽电荷平衡法通过迭代求解原子电荷,实现分子内电负性均衡,例如Au₁₃Naₙ团簇的电荷分布优化。DFT的精度依赖泛函选择和基组优化,但弱相互作用的描述仍存在挑战。
对于重元素,需引入相对论效应进行高阶校正,采用Dirac-Coulomb-Breit哈密顿量结合CCSD (T) 方法计算IP和EA,进而得到χ=(IP+EA)/2。以Nh为例,其电负性高于同族Tl,源于相对论导致的电子轨道收缩效应。
当前模拟挑战主要集中在基组不完整、电子相关效应缺失及相对论计算的高成本——DFT的复杂度限制了大体系应用,需通过算法优化提升效率。
这些方法的协同应用,从分子轨道近似到严格量子力学框架,构建了从有机小分子到重元素体系的电负性模拟体系,为化学键极性分析、材料电子结构设计及复杂体系反应机理研究提供了精确的理论工具,推动电负性概念从经验尺度向量子力学精确描述的深度演进。
DOI:10.56042/ijc.v63i1.1093
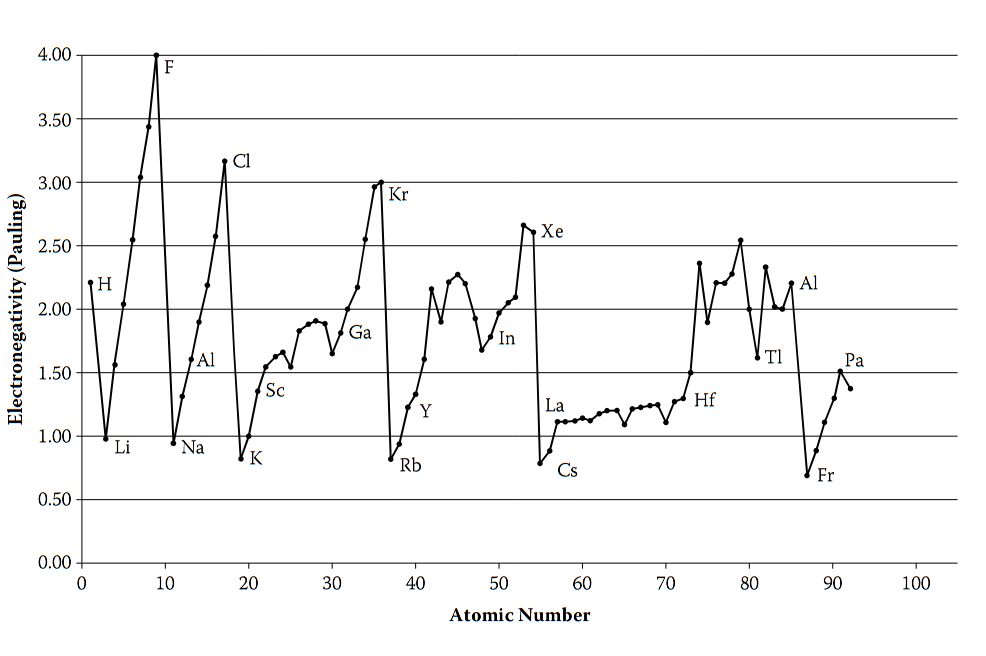
电负性尺度周期性
Boeyens构建的电负性周期性图表,以原子序数为横轴、归一化电负性值为纵轴,对比了Pauling、Allred和AR尺度的电负性分布,并关联带隙能量,系统揭示了电负性的周期性演化规律。
该图表显示,元素电负性呈现同周期从左到右递增、同族从上到下递减的典型周期性特征;值得注意的是,Pauling尺度在过渡金属区出现数值异常,反映出经验模型对d轨道电子相关性的描述局限,而隐含于带隙关联的Allen尺度表现更平滑,与量子力学预期一致。
图表验证了电负性作为“周期表第三维” 的科学价值,其周期性变化精准解释了元素化学行为的突变,而Allen模型因将电负性与价电子能量直接关联,为这种周期性提供了物理一致性的理论基础,架起了原子电子结构与宏观化学性质的桥梁。
DOI:10.1038/s41467-021-22429-0
超重元素电负性计算的应用
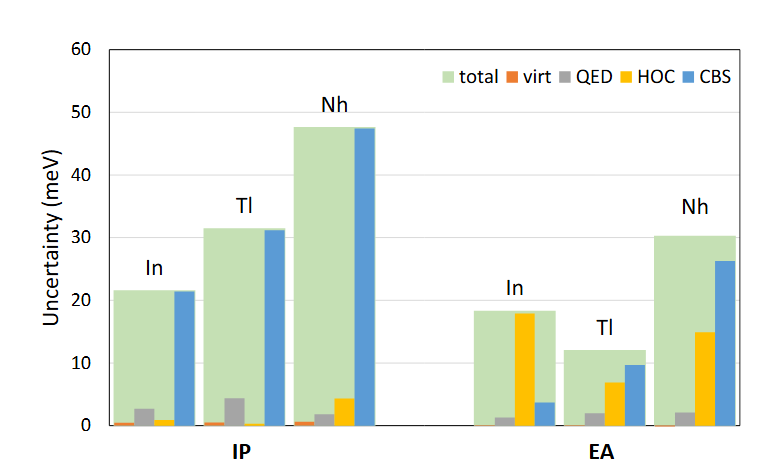
Guo等人在高被引论文中针对超重元素Nh开展的电负性计算研究,为极端条件下元素性质的理论预测提供了典范。
作为实验合成极具挑战性的超重元素,Nh的化学性质研究高度依赖理论计算,该工作采用相对论耦合簇方法,在包含Dirac-Coulomb-Breit项的哈密顿量框架下,系统探究了其电子亲和能(EA)与电离能(IP),进而推导电负性参数。
计算流程首先通过求解Kohn-Sham方程获得初始电子密度ρ(r),在此基础上通过精确计算能量差,结合Helgaker基组外推方案与量子电动力学效应校正,最终得到Nh的电负性,这一数值显著高于同族元素Tl,其物理根源在于相对论效应导致的7s轨道收缩——该效应增强了原子核对外层电子的吸引能力,使Nh对电子的束缚力提升。
电荷分布分析进一步表明,Nh的价电子呈现显著局域化特征,这一特性与其高反应活性直接相关,为理解超重元素的化学行为提供了电子结构层面的解释。
该研究的理论贡献在于,通过DFT与高阶电子相关校正的协同应用,将电负性预测误差控制在,验证了量子化学方法在超重元素研究中的可靠性。
具体而言,相对论耦合簇方法对电子相关效应的精确描述,结合Breit项对磁相互作用的处理,克服了传统密度泛函理论在强相对论体系中的局限性,为超重元素的电子结构计算建立了基准方法。
从应用价值看,该工作证实电负性模型在极端物理条件下的预测能力,其提供的参数为设计含超重元素的功能材料提供了关键电子结构参数,推动了计算化学在核素研究与材料设计交叉领域的应用。
这一案例同时揭示了理论化学在突破实验限制、拓展元素周期表认知边界中的独特价值——通过多尺度量子力学计算与高精度校正策略,可实现对难以合成元素的性质预测,为探索物质世界的极限特性开辟了理论路径。
DOI:10.1088/1361-6455/ac761f
总结
电负性作为衔接微观电子结构与宏观化学性质的核心参数,其理论计算已构建起从经验近似到量子力学严格描述的多尺度框架。
早期Pauling基于热化学键能差异建立的经验模型,经Mulliken整合电离能与电子亲和能的绝对能量尺度及Allen基于价电子光谱数据的高精度模型,逐步发展至DFT框架下以化学势定义的量子力学严格表述,实现了从定性描述到定量计算的精度跃升。
计算方法的革新同步推进:HOMO-LUMO轨道能量法为分子电负性提供高效筛选工具,DFT通过自洽电荷均衡实现复杂体系的自适应模拟,而相对论耦合簇方法则攻克了超重元素电负性计算的难题。
这些理论进展已深度拓展至材料科学与催化领域,包括超导材料带隙预测、电负性–反应选择性关联分析及纳米金刚石固氮反应性调控等关键应用。
电负性理论研究将聚焦于发展机器学习加速的DFT方法以降低大体系计算成本,整合电负性与化学硬度构建全局反应描述符以深化对化学反应路径的量化理解,以及探索非平衡态下激发态动力学过程中的电负性演化规律。
通过系统梳理电负性的理论本质、计算方法及应用场景,为计算化学研究提供了从基础理论到前沿应用的完整认知框架,助力研究者在材料设计、反应机理预测等领域实现基于电负性调控的精准科学设计。