本文系统阐述了如何通过密度泛函理论(DFT)来解释催化反应中的产物选择性问题。首先,介绍了DFT的基本原理及其在催化领域的应用,如吸附能计算、反应能垒评估和电子结构分析。
接着,文中详细讨论了吸附构型和吸附能如何影响反应路径的选择,以及通过构建反应路径和识别过渡态揭示不同产物通道的动力学差异。
最后,本文强调了Gibbs自由能台阶图在可视化热力学和动力学控制机制中的重要性,并通过多个实例展示了DFT计算如何解释实验中观察到的高选择性行为,体现其在催化剂设计与反应机理研究中的关键作用。
催化反应中产物的选择性是实验催化研究的关键关注点之一。
借助密度泛函理论(Density Functional Theory, DFT)的理论计算工具(如VASP软件),我们可以在原子和电子层面探究吸附和反应过程,从而帮助解释实验中观察到的选择性现象,并指导催化剂的设计与机理研究。
下面将从几个方面介绍DFT在催化选择性研究中的作用。
DFT是一种基于量子力学的计算方法,以电子密度而非波函数为基本变量来求解多电子体系的能量。在DFT计算中,体系的电子分布被迭代优化,以满足Kohn-Sham方程,从而得到体系的基态能量和电子结构。
对于固体表面催化研究,常采用平面波赝势方法的软件(如VASP)建立周期性模型,模拟催化剂表面与分子吸附的相互作用。计算结果包括吸附构型、键长键角、体系总能量、电子态密度等信息,可用于分析反应的热力学和动力学性质。
吸附性质预测: 计算反应物、中间体、产物在催化剂表面的吸附能,判断哪些物种更易吸附,以及吸附后的构型和键合情况。这有助于确定反应活性位以及底物在表面的取向,为理解哪些物种更可能参与后续反应提供依据。
反应热力学与动力学: 计算各元素反应步骤的反应能变化(ΔE)和活化能(即过渡态能垒),从热力学角度判断反应自发性,从动力学角度评估反应速率和难易。通过识别最高能垒的步骤,可以预测可能的速率控制步骤。
电子结构分析: 分析催化剂与吸附物的电子结构(如轨道杂化、价态变化、d带中心等),以及键级、电荷分布等,从微观角度解释吸附强度和反应活性的来源。例如,DFT计算提供的电子态密度和差分电荷密度图有助于理解催化剂表面对特定键的活化能力。
总之,DFT理论计算已成为催化研究的重要工具。对于以实验为主的研究者来说,理解DFT提供的这些信息,可以将实验宏观现象与微观机理联系起来,从而更全面地认识催化反应的选择性成因。
在多相催化中,选择性往往从第一步吸附就开始决定。不同产物路径涉及的反应物或中间体可能以不同方式吸附在催化剂表面,而DFT可以揭示这些差异。
通过DFT优化,我们可以得到底物分子在催化剂表面的吸附构型(例如平躺或竖直,哪一个原子与表面结合等)以及对应的吸附能大小。一般而言,吸附能越高(数值越负)表明物种与表面结合越牢固。
选择性吸附会影响后续反应走向:如果某一种中间体在表面特别稳定,它可能抑制其它路径;反之,如果目标产物在生成后与表面作用较弱,则容易脱附而避免进一步转化。
例如,在选择性加氢反应中,人们希望中间产物(如由炔烃加氢得到的烯烃,或由不饱和醛加氢得到的不饱和醇)在表面适度吸附,一旦形成即可脱附,从而避免过度氢化为不需要的产物。DFT计算揭示了催化剂组分的调整如何实现这一点:Pd-Ag合金催化剂就是一例。
纯Pd表面上乙烯的氢化反应与乙烯脱附的能垒相当,因而乙烯生成后容易继续被氢化;而加入Ag后,乙烯在PdAg表面的脱附变得相对更容易(乙烯氢化的活化能提高,高于其脱附能),使乙烯倾向于从表面逸出而不是进一步加氢成乙烷。
这一DFT发现很好地解释了工业上通过向Pd催化剂中添加Ag来提高乙烯选择性的实验策略。
又如,在过氧化氢直接合成反应中,加入卤素离子可以提高H₂O₂选择性而抑制副产水的生成。DFT计算展示了其中机理:以Br⁻促进的Pd催化剂为例,Br原子吸附在Pd表面会产生静电排斥和位阻效应,改变反应物的吸附方式。
具体来说,Br的存在提高了副反应(如O–O键断裂生成水)的能量障碍,同时对主要反应(H₂O₂生成)的影响较小。在Pd(100)晶面上,Br与吸附O原子的排斥使O₂解离的过渡态构型受扰,从而显著提高了O₂解离的能垒,抑制了深度加氢生成水的路径。
另外,在Pd(211)阶梯面上,Br会阻挡催化剂表面的某些活性位点,迫使反应在另一晶面上进行,从而避开易产生活泼氧的途径。结果是,O₂主要以分子态吸附并参与过氧化氢形成,而不是解离成原子氧继续生成水,H₂O₂的选择性因此得到提升。
通过这些DFT分析,我们可以直观理解表面修饰(如加卤素、合金化)如何改变吸附行为,从而调控催化选择性。
DOI: 10.1016/j.cattod.2021.09.030
除了初始吸附,催化反应的选择性还取决于后续每个反应步骤的动力学障碍。DFT计算可以系统地构建完整的反应路径,即从反应物经过一系列中间体和过渡态最终到达不同产物的过程,并为每一步提供相应的能量数据。在实践中,这通常包括以下步骤:
1. 确定反应步骤与中间体: 根据实验线索或化学直觉,假设可能的反应机理,包括各中间体物种和转化步骤。在DFT模型中构建这些物种吸附在催化剂表面的初始构型。
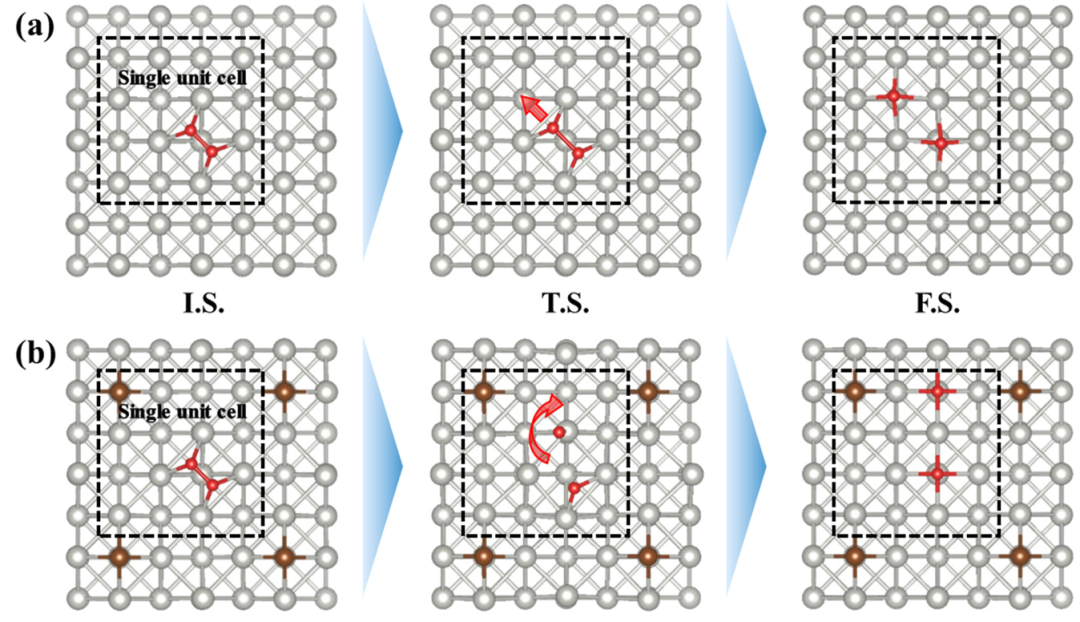
2. 过渡态搜索: 对于每一个假设的反应步骤,利用过渡态搜索方法(如NEB方法或微扰法)来寻找过渡态结构。过渡态是连接反应物和产物的高能量点,其特征是在振动分析中出现唯一的虚频对应于反应坐标方向。
3. 计算反应能垒: 获取过渡态之后,可计算活化能(即过渡态相对于反应物态的能量差)以及反应能(产物态相对于反应物态的能量差)。这些数值反映了该步骤进行的难易程度和热效应。
4. 比较各步能垒: 将一条路径中所有步骤的活化能排列比较,通常最高的那一步就是速率控制步骤。它对应着整个路径中最慢的一环,往往决定了产物生成速率。如果某一步的能垒过高,反应在实际条件下可能根本无法越过这一关卡而停滞。
5. 评估竞争路径: 对不同产物对应的路径重复上述计算。通过对比各路径中的最高能垒和总体能量变化,可以判断哪条路径更有优势。如果产物A路径中的最高能垒低于产物B路径,那么在动力学控制下反应将倾向于选择产物A。
运用上述方法,DFT计算能够揭示选择性背后的动力学原因。
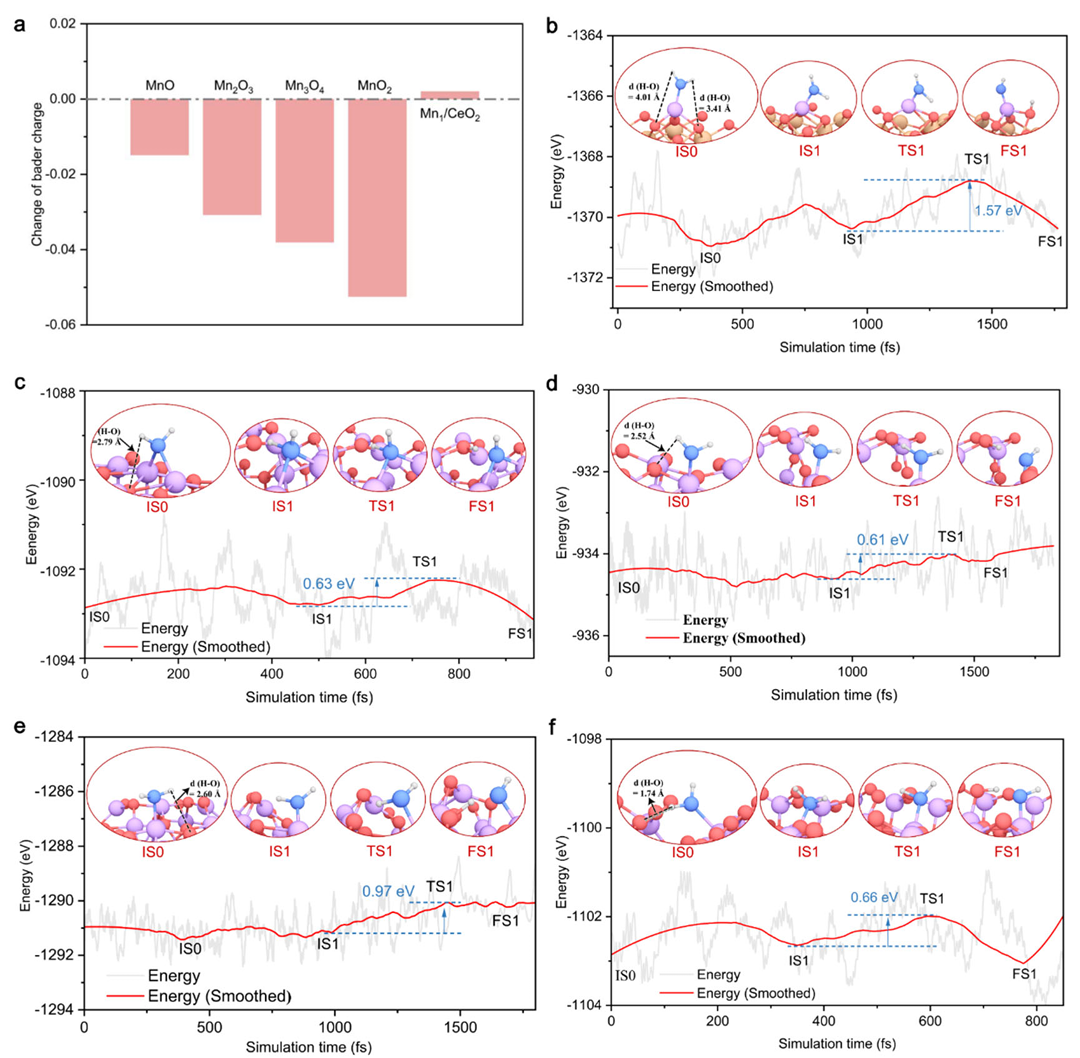
例如,在NH₃选择性还原NOₓ(NH₃-SCR)反应的研究中,针对生成N₂或副产N₂O的两种路径,DFT计算发现:沿N₂O生成的路径存在一个非常高的能垒(超过1.5 eV),对应于关键中间体的转化步骤,这使得该路径在反应中几乎不发生。
相比之下,生成N₂的主要路径各步能垒都相对较低(例如某关键步骤的活化能仅0.75 eV),因此反应主要沿这一低阻力通道进行。在这种情况下,最高能垒大的副反应被“堵住”了,体现为催化剂对N₂产物表现出高选择性。
DOI: 10.1038/s41467-025-55838-6
在考虑了吸附和各步反应能垒之后,仍需进一步评估反应条件下各步骤的自发性。这通常需要用到Gibbs自由能而非仅仅 0 K的电子能。
DFT计算所得的能量可以通过加入零点能校正和热力学统计计算,在给定温度和压力下转化为各物种的Gibbs自由能。将反应路径中每个物态(包括反应物、过渡态、中间体、产物)的Gibbs自由能绘制在统一基准之上,就得到所谓自由能台阶图(free energy diagram)。
这幅图形象地展示了沿反应坐标自由能的起伏变化,如同台阶或能量曲线,提供了关于反应方向和选择性的宝贵直观信息:
若最终产物的自由能低于初始反应物,总反应在热力学上是自发的,意味着在足够长时间或适宜条件下反应倾向于生成该产物(热力学控制)。反之,如果某产物对应的路径总体自由能升高,则需要特殊条件才能发生,或可能被其他更自发的路径竞争掉。
台阶图中每一上升的峰对应一个过渡态,自由能最高的峰代表整个反应过程中需要克服的最大障碍,即总的动力学瓶颈。不同路径的自由能图可以直接比较:哪一路径的最高峰更低,哪一个就更容易发生(动力学控制)。
因此,通过自由能图可以判断在给定温度下反应是动力学控制还是热力学控制,以及主要产物的形成受哪一因素支配。
自由能图还体现了中间体的相对稳定性。如果某中间体的Gibbs自由能非常高,则其难以积累,往往迅速转化或不易形成;而自由能较低的中间体则可能是反应停留的“平坦区域”,有时对应实验上检测到的积累物或休止态。
因此,自由能台阶图能够指示反应可能停留在哪个步骤,以及如何通过改变条件来驱动反应继续进行。
DOI: 10.1038/s41467-025-55838-6
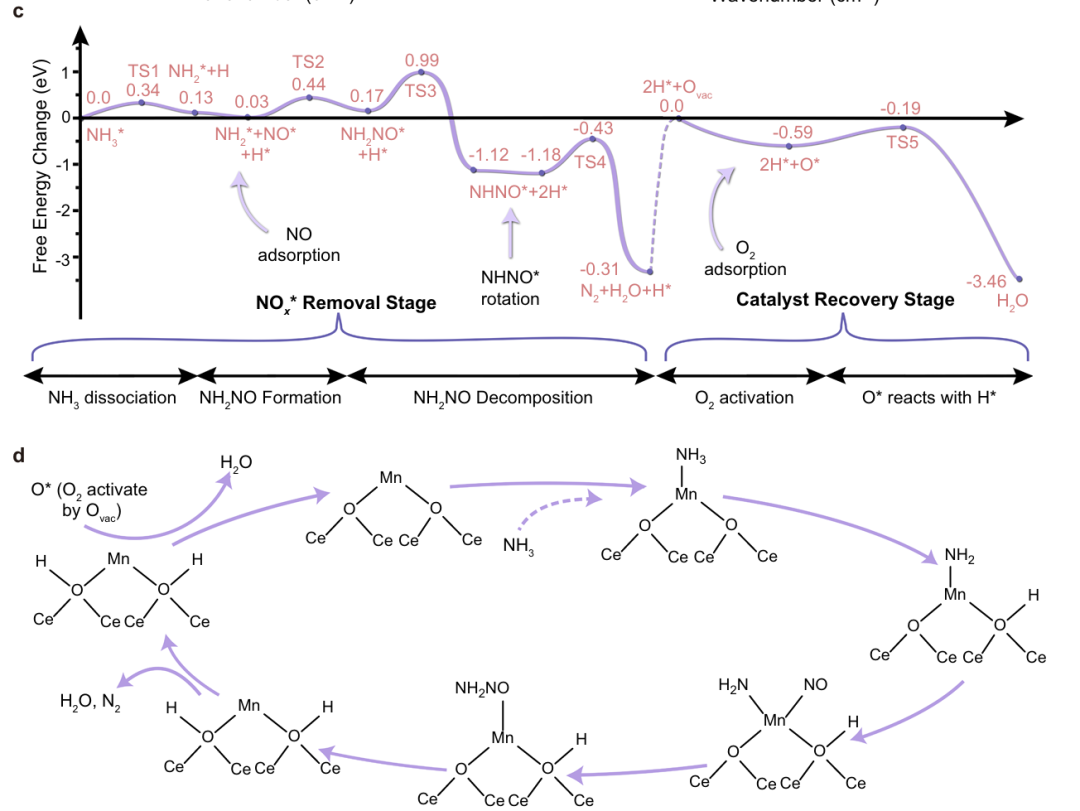
DFT计算得到的NH₃-SCR反应自由能台阶图示例。横轴为反应进程划分的各关键步骤,纵轴表示相对于初始物(NH₃和NO)基准的自由能变化(单位eV)。
从图中可以看出,不同路径的能量障碍有所区别:首先NH₃在催化剂上的活化需要克服一个较小的自由能垒(TS1,约0.34 eV)即可形成中间体NH₂*。之后沿主要路径,中间体经过一系列反应最终生成N₂和H₂O,最高的能垒(TS4)约0.75 eV。
相比之下,副产N₂O的形成涉及的关键步骤TS5具有更高的自由能峰值(~1.56 eV),因此N₂O路径在动力学上受强烈抑制,反应主要朝向生成N₂。
通过这种自由能图的分析,我们直观地理解了哪条路径的能垒最低以及哪种产物在热力学上更稳固,从而解释了催化剂高度选择性地生成N₂而非N₂O。
总而言之,Gibbs自由能台阶图将反应的热力学驱动力和动力学障碍结合展示,使研究者能够一目了然地判断反应更倾向于哪个方向进行以及影响选择性的关键步骤。
这对制定实验策略(如调节温度、压力以控制选择性)和设计催化剂(如稳定特定中间体或降低特定步骤能垒)都有直接的指导意义。
面对复杂的催化反应,实验手段有时难以直接窥探微观机理,而基于DFT的理论计算正好弥补了这一空白。
对于主要从事实验研究的工作者而言,适当利用DFT计算所提供的信息,可以将“黑箱”般的催化过程拆解为可理解的步骤:哪种分子更偏好被表面吸附、关键键如何断裂形成、能垒最高的瓶颈在哪里,以及为何某一产物占优势等等。
这些洞见不仅加深了对现有催化体系的理解,也能启发新的实验思路(例如调整催化剂组成、改变操作条件以提高所需选择性)。
需要强调的是,理论计算的预测往往需要与实验验证相结合:DFT提供的是理想化模型下的趋势和机理,需要通过实验结果加以校正和确认。因此,在实际研究中,应当将DFT计算视作辅助工具,与实验探究相互印证。
总的来说,DFT理论计算已成为催化机理研究中不可或缺的一环,通过桥接原子尺度和实验宏观现象,为实现对催化反应选择性的精确调控提供了强有力的支持。
本次课程基于CASTEP、DMol3模块设计,直击催化科研热点,通过28小时高强度实操培训,用通俗的语言将HER/OER/ORR/CO2RR台阶图、火山图、d带理论、过渡态、带隙工程、能带电位匹配等催化计算核心问题讲透彻,使大家能将DFT计算用到自己的文章中。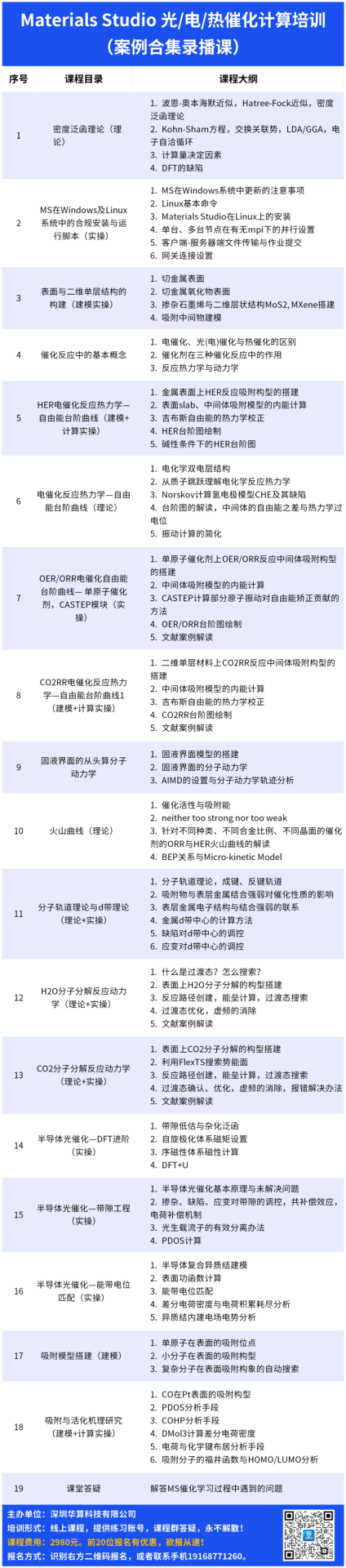

声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!