




HOMO(最高占据分子轨道)和 LUMO(最低未占据分子轨道)是分子轨道理论中的重要概念,在化学、材料科学等领域有着广泛的应用,它们具有如下意义:
1、化学反应活性
亲电和亲核反应:HOMO 轨道上的电子具有较高的能量,容易参与化学反应。在亲电反应中,亲电试剂倾向于进攻分子的 HOMO 轨道,因为这里的电子云密度相对较高,电子更容易被亲电试剂所吸引。而 LUMO 轨道由于能量较低且是空的,在亲核反应中,亲核试剂会将电子给予分子的 LUMO 轨道。
反应的可能性和方向:HOMO 和 LUMO 轨道的能量差(能隙)对反应的可能性和反应方向起着关键作用。一般来说,能隙越小,分子越容易发生化学反应。当两个分子相互作用时,一个分子的 HOMO 和另一个分子的 LUMO 之间的能量匹配程度决定了反应的可行性和选择性。
2、分子的稳定性
能量角度:HOMO 能量较低的分子通常更稳定,因为其电子处于相对较低的能量状态,需要更多的能量才能将电子激发或移除。相反,LUMO 能量较高的分子也更稳定,因为要使其他电子填充到该轨道上需要提供较多的能量。
芳香性和反芳香性:HOMO 和 LUMO 轨道的特征与分子的芳香性和反芳香性密切相关。具有芳香性的分子通常具有特殊的 HOMO – LUMO 分布,满足休克尔规则,即具有 (4n + 2) 个 π 电子的平面环状共轭体系具有较高的稳定性,其 HOMO 和 LUMO 轨道的能量和对称性有助于维持这种稳定结构。而反芳香性分子的 HOMO – LUMO 特征使得它们具有较高的反应活性和较低的稳定性。
3、光学和电学性质
吸收光谱:分子吸收光子后,电子可以从 HOMO 跃迁到 LUMO。吸收光子的能量与 HOMO – LUMO 能隙相对应,因此通过测量分子的吸收光谱,可以得到关于 HOMO – LUMO 能隙的信息。这对于理解分子的颜色、光吸收特性以及设计具有特定光学性质的材料(如染料、发光材料等)非常重要。
导电性:在有机半导体材料中,HOMO 和 LUMO 轨道的离域程度以及它们之间的能隙大小决定了材料的导电性。较小的能隙意味着电子更容易从 HOMO 跃迁到 LUMO,从而在材料中形成电流,因此具有较小 HOMO – LUMO 能隙的分子材料通常具有较好的导电性。
4、分子结构和化学键
轨道重叠和键的形成:HOMO 和 LUMO 轨道的形状和对称性对于理解分子中化学键的形成和分子的几何结构至关重要。分子轨道的重叠程度决定了化学键的强度和方向,而 HOMO 和 LUMO 轨道在这个过程中起着关键作用。
分子间相互作用:分子之间的相互作用(如氢键、范德华力等)也与 HOMO 和 LUMO 轨道有关。一个分子的 HOMO 可以与另一个分子的 LUMO 相互作用,从而影响分子的聚集状态和物理性质。
VASP(Vienna Ab initio Simulation Package)是一款广泛应用于材料科学与量子化学领域的计算软件,用于计算 HOMO(最高占据分子轨道)和 LUMO(最低未占据分子轨道)轨道的流程如下:
1、准备输入文件
需要准备三个主要的输入文件:POSCAR、POTCAR 和 INCAR。
-
POSCAR:此文件用于描述原子的位置与晶胞结构。你可以采用文本编辑器直接编写,或者借助可视化软件(如 VESTA)生成。
-
POTCAR:该文件包含了赝势信息。你要从 VASP 赝势库中选取合适的赝势文件,然后将它们合并成一个 POTCAR 文件。
-
INCAR:此文件用于设置计算参数。以下是一个基本的INCAR 文件示例:

在这个示例里,ENCUT 设定了平面波截止能量;ISMEAR 和 SIGMA 是用于处理电子占据的参数;IBRION 和 NSW 被设置为不进行结构优化;NELM 是最大自洽迭代次数;LWAVE 和 LCHARG 分别控制是否保存波函数和电荷密度。
2、进行自洽场(SCF)计算
在准备好输入文件之后,就可以开展自洽场计算了。在终端中输入如下命令:
mpirun -np 4 vasp_std
这里的mpirun -np 4 表示使用 4 个并行进程,vasp_std 是 VASP 的标准执行程序。
3、分析输出结果
自洽场计算完成后,会生成一个EIGENVAL 文件,该文件包含了所有的能级信息。你可以使用 Python 脚本读取并分析这个文件,以下是一个简单的 Python 脚本示例:
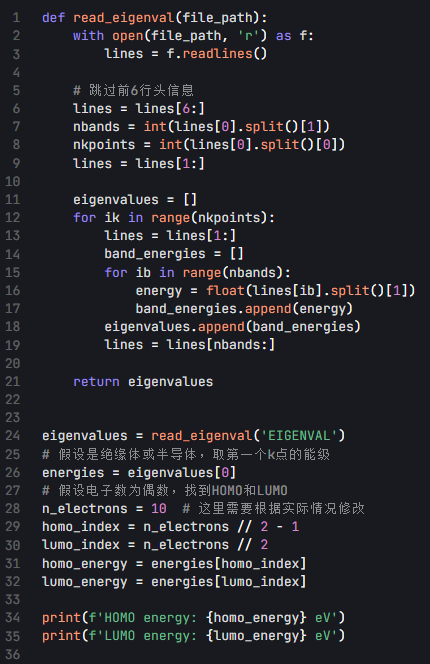
这个脚本会读取EIGENVAL 文件,提取能级信息,然后根据电子数确定 HOMO 和 LUMO 的能量。
4、注意事项
赝势选择:要依据具体的元素和计算需求来选择合适的赝势。
计算精度:可以通过调整ENCUT、ISMEAR 和 SIGMA 等参数来提高计算精度。
电子数:在分析EIGENVAL 文件时,要准确确定电子数,这样才能正确找到 HOMO 和 LUMO。
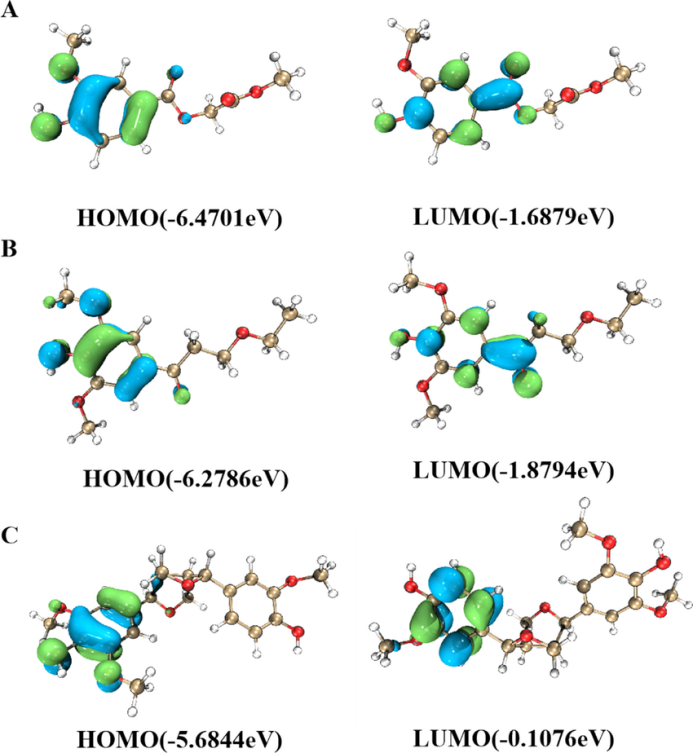
HOMO/LUMO能级位置与轨道分布
通过密度泛函理论(DFT)计算得到的 HOMO 和 LUMO 结果,可以从以下几个方面进行分析:
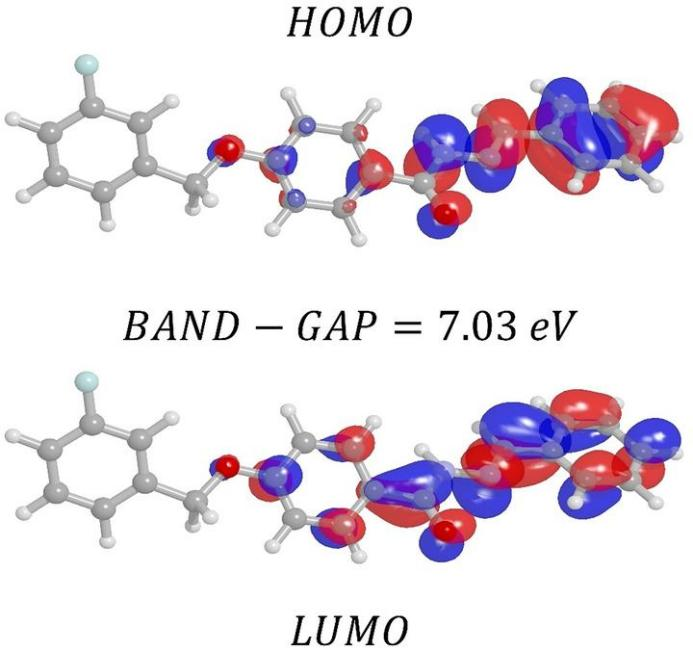
轨道能量差,即HOMO 和 LUMO 的能量差ΔE。它是是一个关键指标,较小的能量差意味着电子从 HOMO 跃迁到 LUMO 相对容易,分子可能具有较好的光学和电子性质,如在光电器件中表现出较高的光电转换效率。例如,对于有机半导体材料,较小的能隙有利于吸收可见光,从而产生更多的激子。
绝对能量值,即HOMO/LUMO绝对位置。HOMO 能量越高,电子越容易被激发,分子的供电子能力可能越强;LUMO 能量越低,分子接受电子的能力越强。在研究分子间的电荷转移时,HOMO 和 LUMO 的绝对能量值可以帮助判断电子转移的方向和趋势。如果一个分子的 HOMO 能量高于另一个分子的 LUMO 能量,那么在适当条件下,电子可能从前者转移到后者。
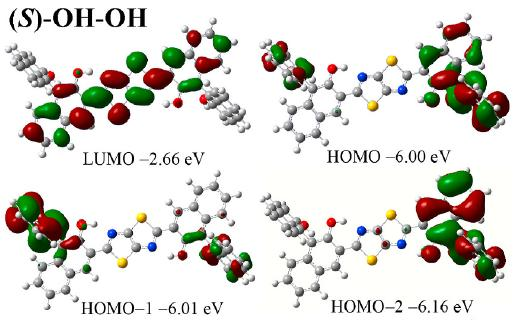
轨道成键与反键特征,即电子云分布情况。通过轨道形状可以判断 HOMO 和 LUMO 是成键轨道还是反键轨道。成键轨道的电子云在原子核之间分布较多,对分子的稳定性有积极作用;反键轨道的电子云则在原子核之间分布较少,且有节点,会削弱分子的稳定性。例如,在双原子分子中,HOMO 可能是成键轨道,而 LUMO 是反键轨道。当分子吸收能量发生电子跃迁时,电子从成键的 HOMO 跃迁到反键的 LUMO,分子的稳定性会暂时降低。
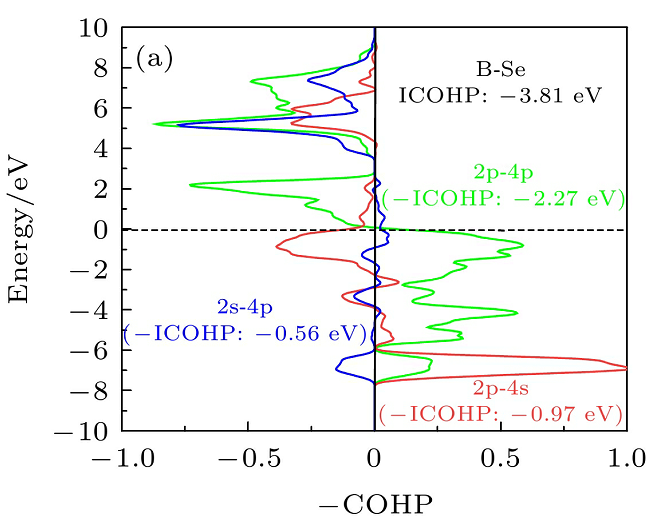
原子轨道贡献,即电子云在各个原子上的分布情况。这体现了不同原子对 HOMO 和 LUMO 的贡献程度。如果某个原子在 HOMO 上的电子云密度较大,说明该原子在提供电子方面起主要作用;而在 LUMO 上电子云密度较大的原子,则更倾向于接受电子。比如在一些有机金属配合物中,通过分析 HOMO 和 LUMO 的电子云分布,可以确定金属原子和配体之间的电子转移情况,进而理解配合物的化学性质和反应活性。
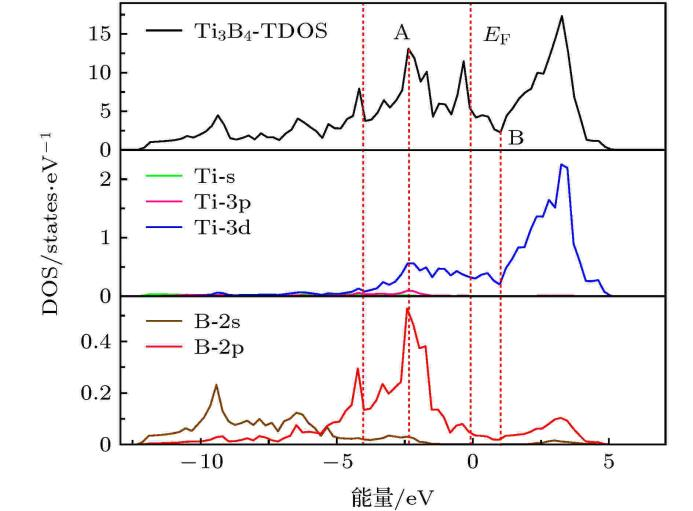
亲电与亲核反应位点,可判断不同反应的类型。HOMO 电子云分布集中的区域通常是亲电反应可能发生的位点,因为这些区域电子云密度高,容易被亲电试剂进攻;而 LUMO 电子云分布集中的区域则是亲核反应的潜在位点,容易接纳亲核试剂的电子对。例如,在一个含有多个官能团的有机分子中,通过分析 HOMO 和 LUMO 的电子云分布,可以预测亲电试剂更可能进攻哪个官能团,以及亲核反应的优先发生位置。
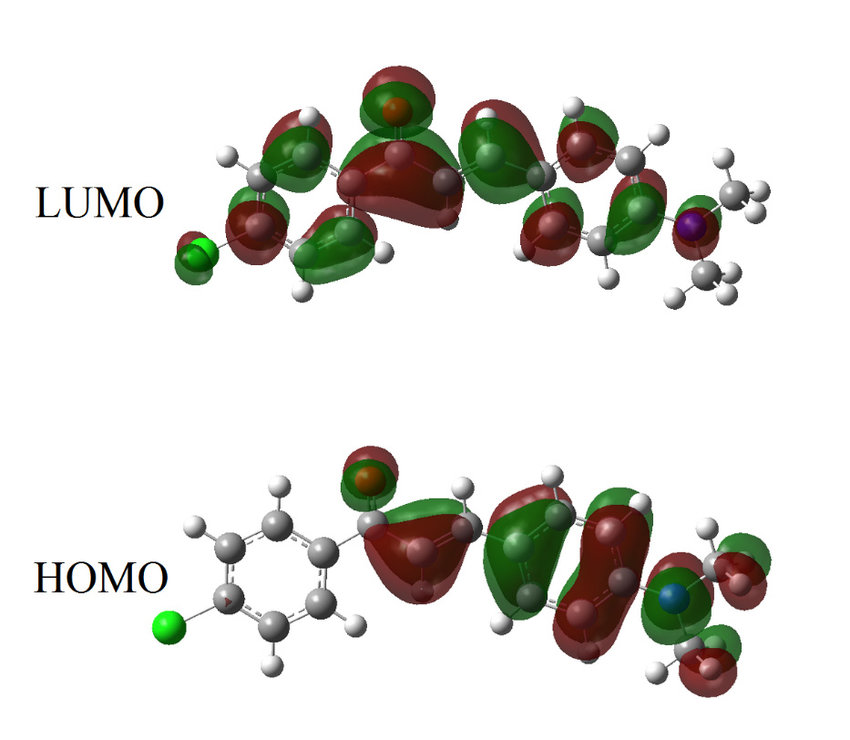
反应机理研究,即得失电子的方向。结合反应条件和实验现象,HOMO 和 LUMO 的计算结果可以帮助推测化学反应的机理。例如,在研究光催化反应时,通过分析反应物和催化剂的 HOMO 和 LUMO,能够理解光激发下电子的转移路径和反应中间体的形成过程,从而为优化光催化反应提供理论依据。