

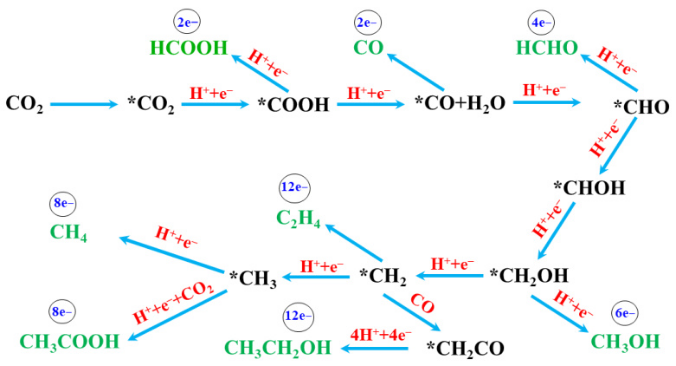
CO₂RR机理涵盖电催化与光催化。电催化中,CO₂在催化剂表面吸附、活化,经中间体转化生成产物;光催化则涉及多种反应机制,如甲醛、卡宾等路径。不同产物对应不同路径,如CO生成路径为CO₂→*CO₂→*COOH→*CO→CO↑,甲酸路径为CO₂→*CO₂→*HCOO→HCOOH。
DFT计算在CO₂RR机理研究中应用广泛,可解析反应路径、确定活性位点、优化催化剂性能、研究反应动力学,并分析产物选择性。
CO2RR反应机理概况
二氧化碳还原反应(CO₂RR)是一种将CO₂转化为高附加值化学品或燃料(如CO、甲酸、甲烷、甲醇、乙烯等)的电催化过程,其基本机理涉及多个步骤和路径。CO₂RR通常包括以下关键步骤:
CO₂吸附:CO₂从电解质中转移到电极表面,形成吸附态分子(如*CO₂)。这一步是反应的起始步骤,吸附能的大小直接影响后续反应的活性和选择性。
中间体生成:吸附态的CO₂通过电子转移或质子耦合反应生成关键中间体,如*CO、*CHOH等。这些中间体是决定最终产物的重要因素。例如,*CO的形成是许多C1产物(如CO、甲酸盐)生成的关键步骤。
C-C键形成:在某些路径中,中间体进一步反应生成C-C键,从而形成多碳产物(如乙烯、乙醇等)。这一步骤通常需要较高的过电位,并且受到催化剂表面性质的影响。
脱附与产物释放:最终产物从催化剂表面脱附进入电解质溶液中。脱附能的大小决定了产物的选择性和反应的效率。

https://doi.org/10.3390/nano12142379
不同产物反应路径与文献支持
CO(一氧化碳)路径:CO₂→ *COOH → *CO → CO(脱附)。

J. Am. Chem. Soc. 2019, 141, 32, 12717–12723
图中比较了三种不同活性位点(Cu–N₄、Ni–N₄和吡啶氮掺杂的石烯)上CO₂还原为*CO的自由能变化。对于Cu–N₄位点,CO脱附步骤的自由能略微为正(0.12 eV),表明这是一个吸能过程,CO不易从催化剂表面脱附,而是倾向于进一步还原为甲醇。
相比之下,Ni–N₄和吡啶氮位点的*CO脱附自由能为负值(分别为-0.21 eV和-0.54 eV),说明*CO容易脱附生成CO产物。
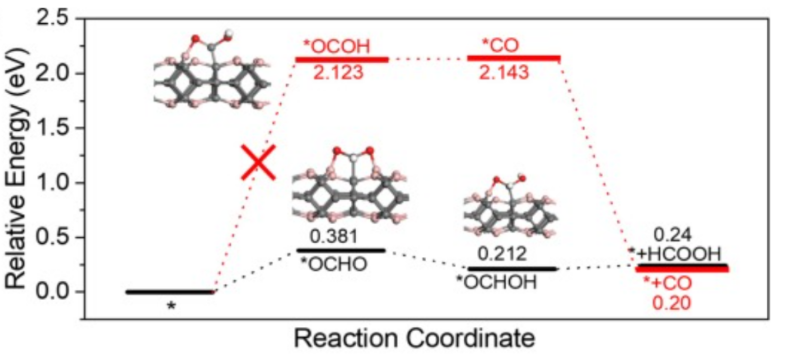
甲酸(HCOOH)路径:CO₂→ *OCHO → HCOOH。
https://doi.org/10.1016/j.jcis.2023.10.058

DOI:10.1002/anie.202311223
图展示了两种铋基沸石有机框架(Bi-ZMOFs)——PZH-1和PZH-2在电催化CO₂还原为甲酸(HCOOH)过程中的自由能变化对比。主要内容如下:
PZH-1(红色曲线):形成关键中间体*OCHO的自由能较低(1.01 eV),表明其更容易生成甲酸。PZH-2(蓝色曲线):生成*OCHO的自由能较高(2.25 eV),导致甲酸生成效率显著低于PZH-1。
关键发现:PZH-1的高选择性(91%法拉第效率)归因于其独特的Bi-N配位结构,促进了电荷从配体N转移到Bi中心,进而稳定*OCHO中间体。理论计算证实,Bi-N配位通过电子转移降低了*OCHO形成的能垒,从而提升了甲酸生成的催化活性。
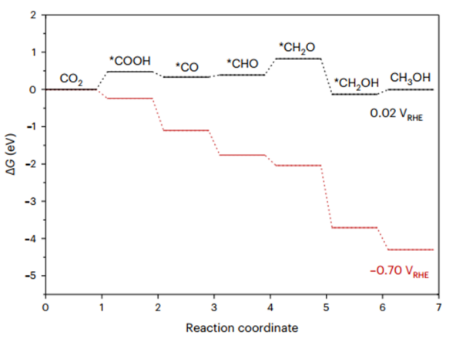
甲醇(CH₃OH)路径:CO₂→ *COOH → *CO → *COH → *CH₂O → CH₃OH。
https://doi.org/10.1038/s41929-024-01197-2

https://doi.org/10.1021/jacs.2c12006
图展示了Cu(111)和La₅Cu₅₅(111)表面在CO₂电还原为甲烷(CH₄)过程中的吉布斯自由能变化对比,主要内容如下:
自由能图对比:La₅Cu₅₅(111)(红色曲线):关键中间体COOH和CHO的形成步骤为放热过程,表明其更容易生成甲烷。*CH₃O倾向于断裂C-O键生成CH₄(放热路径),而进一步氢化为CH₃OH则需吸热。
Cu(111)(蓝色曲线):*CH₃O更倾向于氢化为CH₃OH(放热路径),生成CH₄需克服更高能垒。
关键发现:La的引入通过增强*CH₃O的吸附(形成稳定的La-O键)并促进C-O键断裂,显著降低了甲烷生成的能垒。理论计算与实验(如原位红外光谱)结合,证实La₅Cu₅₅表面更易通过*CO → *CHO → *CH₃O → CH₄路径高效生成甲烷。
甲烷(CH₄)路径:CO₂→ *COOH → *CO → *CHO → *CH₂→ CH₄。

https://doi.org/10.1002/ange.202314121
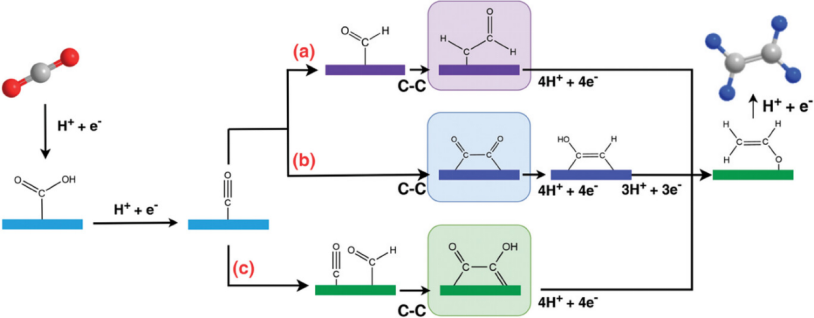
C₂+产物(如乙烯、乙醇)路径:CO₂→ *CO → *CO-CO → *C₂中间体→ C₂H₄/C₂H₅OH。
乙烯
Energy Environ. Sci. 2010, 3, 1311.
Nat. Catal. 2020, 3, 478;
Nat. Commun. 2022, 13, 1877.
在各类涉及eCO2RR制乙烯的研究中,研究者们报道了三种与C-C偶联相关的反应路径。
总之,eCO2RR生产乙烯的关键取决于中间产物的结合能,而提高催化剂表面对*CO的吸附已被证明是促进CO2转化为乙烯的有效方法之一。通过提高*CO的吸附性,可降低二聚机制的障碍,从而提高乙烯的选择性。
乙醇
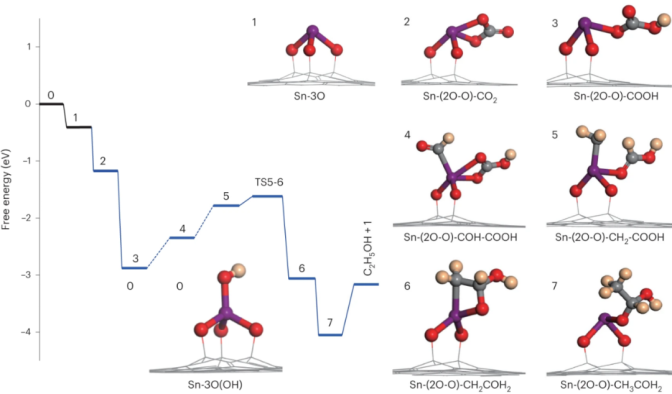
https://doi.org/10.1038/s41560-023-01389-3
通过甲酰基-碳酸氢盐偶联途径的C–C键形成。在Sn1-O3G催化剂上通过CO2RR生成乙醇产物的反应能以及相应的中间体结构
DFT在CO2RR机理研究中的现状
DFT计算在CO₂还原反应(CO₂RR)机理研究中发挥着重要作用,主要体现在以下几个方面:

DOI: 10.1002/bte2.20220012
深入解析反应路径
确定关键中间体:通过计算不同反应路径的吉布斯自由能,确定CO₂还原反应中可能出现的关键中间体。如在CO₂还原生成甲烷的反应中,通过DFT计算明确了许多中间体的结构和能量,从而揭示了反应过程中中间体的转化顺序。
明确反应步骤顺序:DFT计算能够详细描绘CO₂RR的反应路径,包括CO₂的吸附、活化、中间体的形成与转化以及产物的脱附等各个步骤,从而明确各个反应步骤的能垒和反应顺序。
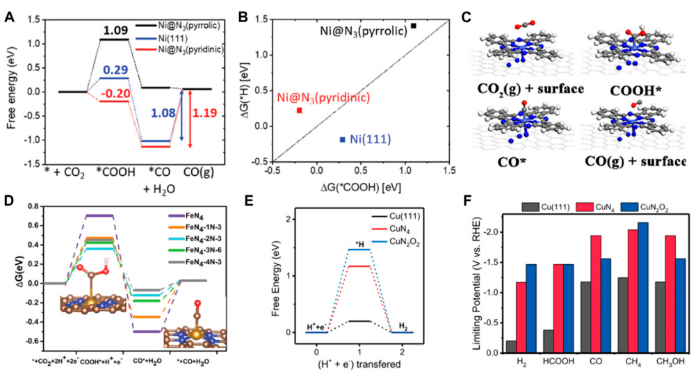
https://doi.org/10.3389/fchem.2023.1172146
确定活性位点
揭示活性位点结构:DFT计算可用于研究催化剂表面不同位点的吸附能和反应活性,从而确定CO₂RR的活性位点。例如,在铜催化剂上,通过DFT计算发现配位不饱和的Cu位点是CO₂RR的活性位点,这些位点能够促进C-C偶联生成多碳产物。
分析活性位点作用机制:通过计算活性位点与反应物及中间体之间的相互作用,揭示活性位点的作用机制。如在氮配位石墨烯负载的三金属单簇催化剂中,DFT计算表明金属与氮之间的电子转移机制可降低CO₂能级,增强CO₂吸附强度,从而提高催化剂的活性。
催化剂设计与优化
指导催化剂设计:基于DFT计算结果,为设计新型高效的CO₂RR催化剂提供理论依据。例如,通过计算不同金属团簇催化剂的电子结构和吸附能,发现某些金属团簇对CO₂具有较强的吸附能力和较低的反应能垒,可作为潜在的高效催化剂。
优化催化剂性能:通过模拟不同反应条件下的反应性能,为优化催化剂的组成、结构和反应条件提供指导。例如,在研究铜基催化剂时,发现通过调节催化剂的表面形貌和组成,可以改变反应中间体的吸附能和反应路径,从而提高催化剂的选择性和活性。
反应动力学研究
计算反应速率:结合DFT计算得到的活化能等参数,利用阿伦尼乌斯方程等动力学模型,预测不同催化剂和反应条件下CO₂RR的反应速率,为优化反应条件提供理论依据。
分析决速步骤:确定CO₂RR中的决速步骤,为提高反应速率提供关键信息。例如,在光催化CO₂还原为CO的过程中,通过DFT计算发现CO脱附是潜在的决速步骤,进而可针对该步骤进行催化剂优化。
理解反应机理与选择性
揭示反应机理:DFT计算能够从原子和分子水平上揭示CO₂RR的反应机理,包括电子转移、质子转移和化学键的形成与断裂等过程。如在研究锰二亚胺催化剂将CO₂转化为CO的反应中,通过DFT计算详细解析了反应过程中电子转移和化学键变化的细节。
分析产物选择性:通过计算不同产物生成路径的自由能变化和活化能,预测CO₂RR的产物分布,从而为提高目标产物的选择性提供指导。例如,在CO₂还原生成多碳产物的研究中,DFT计算表明C-C偶联步骤的能垒是影响多碳产物选择性的关键因素。
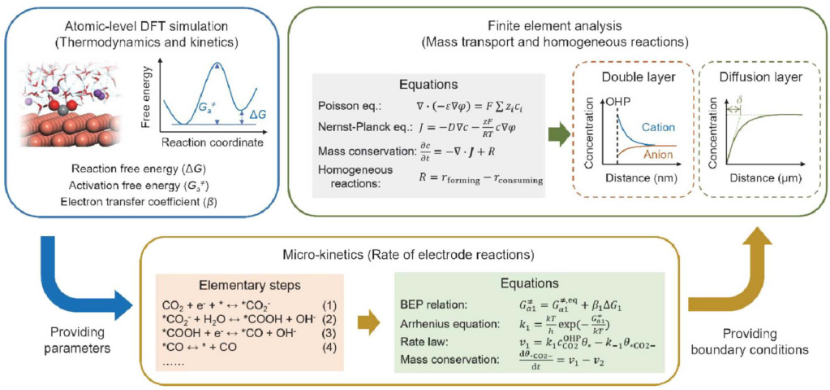
X. Y. Zou and J. Gu, Chin. J. Catal., 2023, 52, 14–31.
总结
CO2RR机理研究高度依赖DFT计算,但其需结合实验验证和更精确的理论方法以克服系统性误差,尤其在C₂+产物生成和表面覆盖度建模方面仍需突破。