

本文深入探讨了锂硫电池中硫还原反应(SRR)的复杂机理,从S₈到Li₂S的多步转化过程涉及多种中间产物(如Li₂Sₙ),其反应路径直接影响电池的容量、循环稳定性和倍率性能。通过热力学分析(如吉布斯自由能计算)和动力学研究,揭示了SRR反应的关键限制因素,如液–固相变的能垒问题。
文章还以C₅N材料为例,结合DFT计算,定量分析了其对多硫化物的吸附能力、电子结构调控作用以及降低Li₂S分解能垒的机制,为优化锂硫电池阴极材料提供了理论依据。这些研究不仅阐明了SRR反应的本质,还为高性能锂硫电池的设计与开发指明了方向。
SRR反应过程
SRR反应是一个复杂的16电子多步转化过程,从初始的S8到最终的Li2S,涉及多个中间产物和反应步骤。在放电过程中,首先,S8分子在电极表面被锂离子进攻,发生环–链转变,形成较长链的多硫化物离子Li2Sn(n = 8, 6, 4等),这一过程对应于放电曲线的高电压平台(2.4 – 2.1V) 。
随着反应的进行,长链多硫化物进一步被还原为短链多硫化物,最终生成不溶性的Li2S2和Li2S,这一阶段对应于放电曲线的低电压平台(2.1 – 1.8V)。在这个过程中,会产生多种中间体,如Li2S8、Li2S6、Li2S4等,这些中间体的稳定性和反应活性对 SRR 反应的速率和路径有着重要影响。
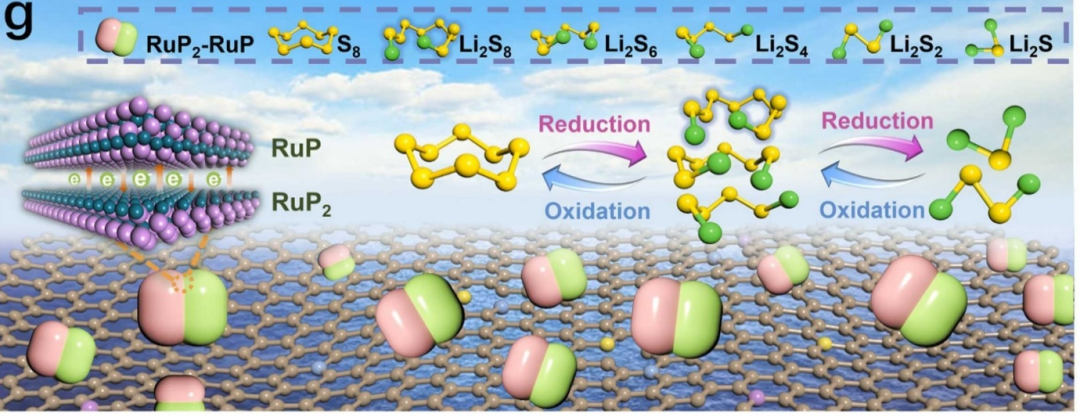

充电过程则是放电过程的逆反应,Li2S和Li2S2被氧化为多硫化物,最终重新生成S8。然而,由于反应动力学的限制以及中间产物的复杂性,充电过程中往往存在较大的过电位,导致能量效率降低。而且,在实际电池运行过程中,SRR反应还受到电极材料、电解质、温度等多种因素的影响,使得反应过程更加复杂。
对电池性能的影响
SRR反应对锂硫电池的性能有着至关重要的影响。首先,它直接决定了电池的容量。由于SRR反应是锂硫电池的核心反应,其反应的完全程度直接影响到硫的利用率,进而决定了电池的实际容量。如果SRR反应能够高效进行,硫能够充分转化为Li2S,则电池可以获得较高的容量。反之,若反应动力学迟缓,部分硫无法参与反应,电池容量将显著降低。
其次,SRR反应的稳定性和可逆性对电池的循环稳定性有着关键作用。在循环过程中,SRR反应的每一步都需要能够稳定且可逆地进行,才能保证电池性能的稳定。然而,由于多硫化物的穿梭效应以及反应中间体的复杂性,SRR反应在循环过程中容易出现不可逆的变化,导致活性物质的损失和电池性能的衰退。
例如,多硫化物在电解液中的溶解和穿梭会导致活性硫逐渐从正极流失,沉积在负极表面,形成 “硫钝化层”,阻碍锂离子的传输和反应的进行,从而降低电池的循环寿命。
此外,SRR反应的动力学性能还影响着电池的倍率性能。在高电流密度下,快速的SRR反应动力学能够保证电池在短时间内进行大量的电荷转移,从而实现高倍率充放电。相反,如果SRR反应动力学迟缓,电池在高电流密度下将无法及时响应,导致电压极化增大,容量迅速衰减,倍率性能变差。
反应路径的热力学分析
反应路径的热力学分析是理解SRR反应能否自发进行以及反应趋势的重要手段,而吉布斯自由能(ΔG)是衡量反应热力学可行性的关键参数。通过 DFT 计算,可以精确地确定SRR反应中各反应物、中间体和产物的吉布斯自由能,从而深入分析反应路径的热力学性质。
以Li2S4还原为Li2S的反应为例,首先计算出Li2S4、Li2S2、Li2S以及反应过程中可能出现的中间体的吉布斯自由能 。根据反应方程式Li2S4 + 2Li+ + 2e–→2Li2S2和Li2S2 + 2Li+ + 2e–→2Li2S,分别计算这两个反应步骤的吉布斯自由能变化(ΔG1和ΔG2)。如果ΔG1 且 ΔG2 ,说明这两个反应步骤在热力学上都是自发进行的,整个反应路径是热力学可行的 。
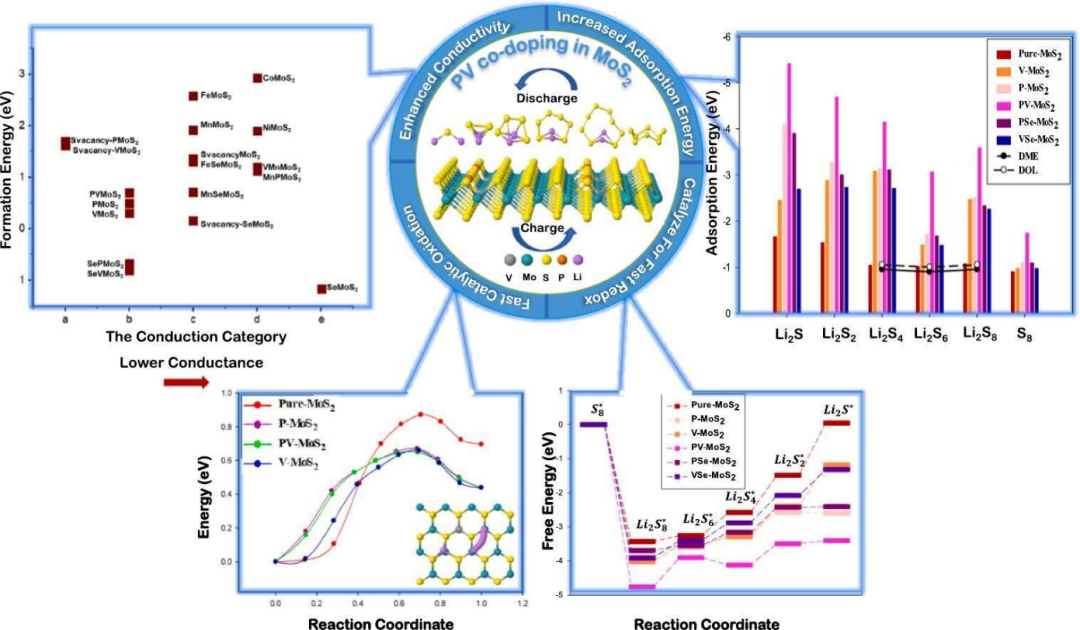
然而,反应的热力学可行性并不等同于反应能够快速发生,还需要考虑反应的动力学因素。而且,反应条件的改变,如温度、压力等,会对反应的吉布斯自由能产生影响,进而影响反应路径的热力学可行性。
通过DFT 计算不同温度下的吉布斯自由能,可以了解温度对反应路径的影响。随着温度的升高,一些反应步骤的吉布斯自由能变化可能会发生改变,原本热力学可行的反应路径可能变得不再有利,或者原本不利的反应路径可能变得可行 。这为优化锂硫电池的工作温度提供了理论依据。
此外,电极材料和电解液的性质也会对反应路径的热力学产生影响。不同的电极材料对多硫化物的吸附作用不同,会改变反应体系的能量状态,从而影响反应路径的吉布斯自由能。一些具有高吸附能的电极材料可以降低多硫化物在电极表面的自由能,使得反应更容易向生成最终产物Li2S的方向进行 。
电解液中的溶剂化作用也会影响多硫化物的稳定性和反应活性,进而影响反应路径的热力学性质 。通过 DFT 计算不同电解液环境下的反应路径吉布斯自由能,可以为电解液的优化设计提供指导。
案例分析
在锂硫电池(LSBs)的研究中,硫还原反应(SRR)是决定电池性能的核心电化学过程,涉及硫(S₈)逐步转化为一系列锂多硫化物(Li₂Sn,2≤n≤8)并最终生成Li₂S的复杂反应路径。本文通过密度泛函理论(DFT)等理论计算方法对SRR进行系统研究,具有重要的理论价值和实践意义,为LSBs的性能优化提供了关键的机理阐释和材料设计指导。
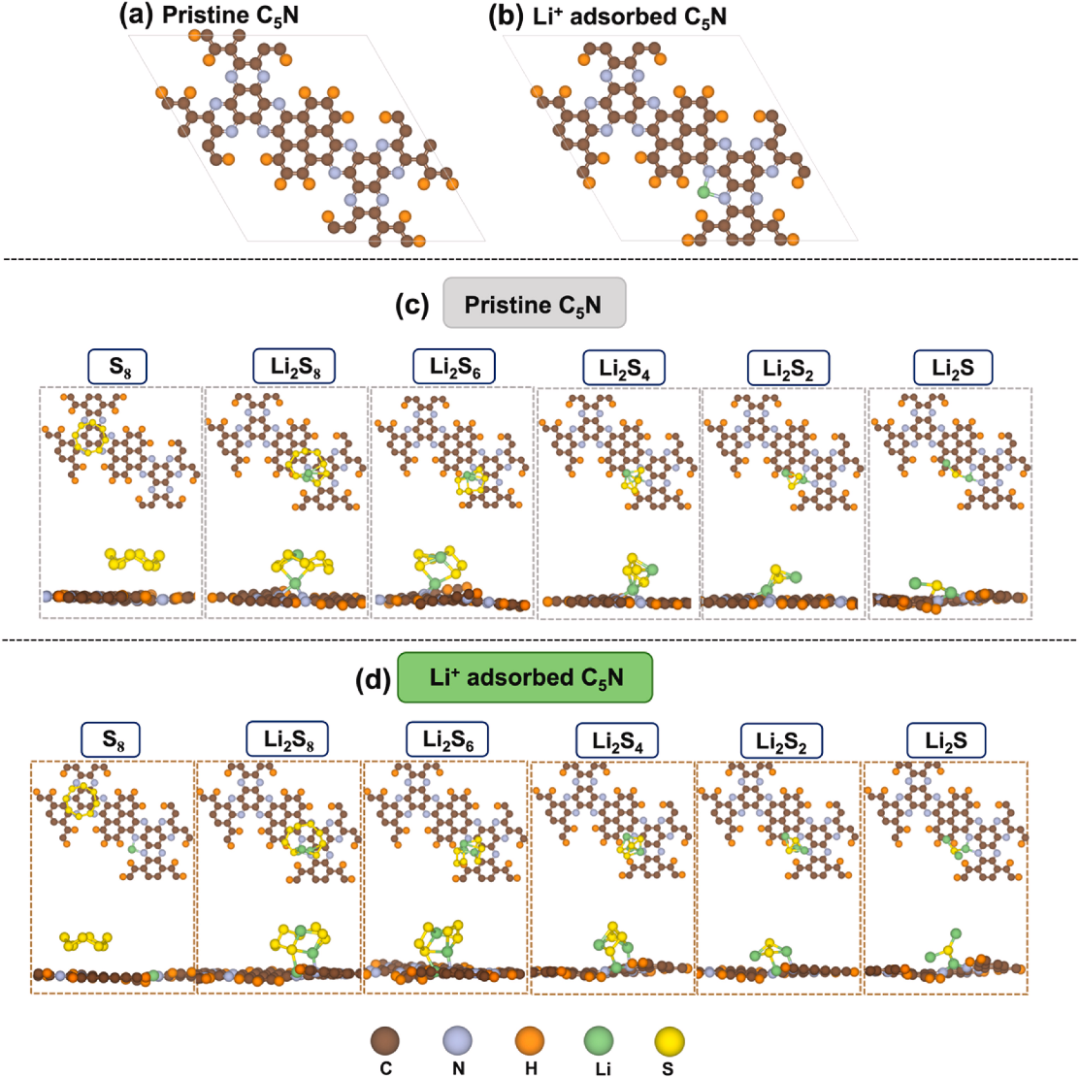
从反应机理层面,理论计算明确了SRR的动力学限制因素。研究表明,SRR过程中的液–液和液–固相变(如Li₂S₄向Li₂S₂的转化)是反应的决速步骤。
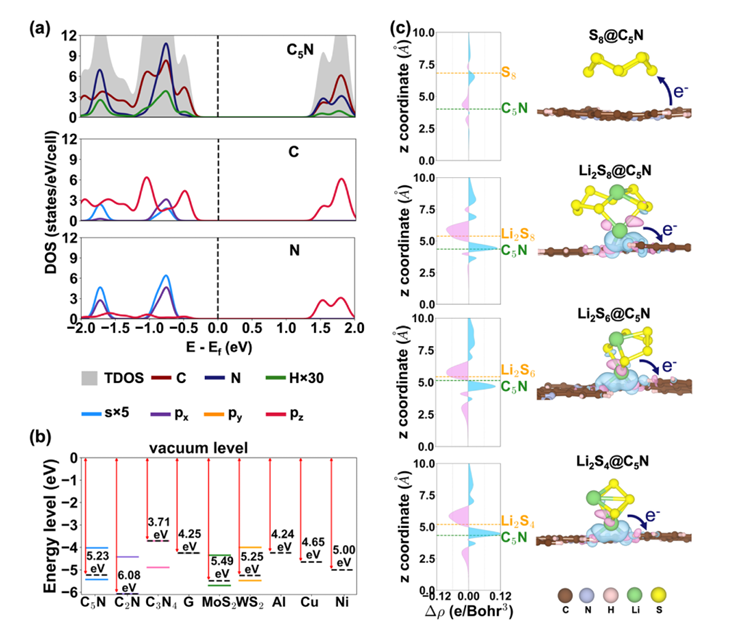
通过计算各反应步骤的吉布斯自由能变(ΔG),发现C₅N作为阴极宿主材料时,Li₂S₄→Li₂S₂的转化能垒仅为0.71 eV,显著低于C₂N(0.92 eV)和N掺杂碳基质(1.02 eV)等其他二维材料。这一结果不仅揭示了C₅N提升SRR效率的本质原因,还为筛选高效催化材料提供了量化依据。
在抑制多硫化物穿梭效应方面,理论计算定量分析了材料与多硫化物的相互作用机制。吸附能(Eₐds)计算表明,C₅N对高阶多硫化物(Li₂Sn,4≤n≤8)的化学吸附能(0.55-1.90 eV)显著高于电解液(如DME、DOL),且主要通过Li-N化学键实现稳定结合,从而有效抑制多硫化物的溶解和迁移。值得注意的是,C₅N具有优化的N/C比例,避免了类似C₃N₄等材料因结合能过强导致的反应中间体“滞留“问题,确保了多硫化物的高效转化。
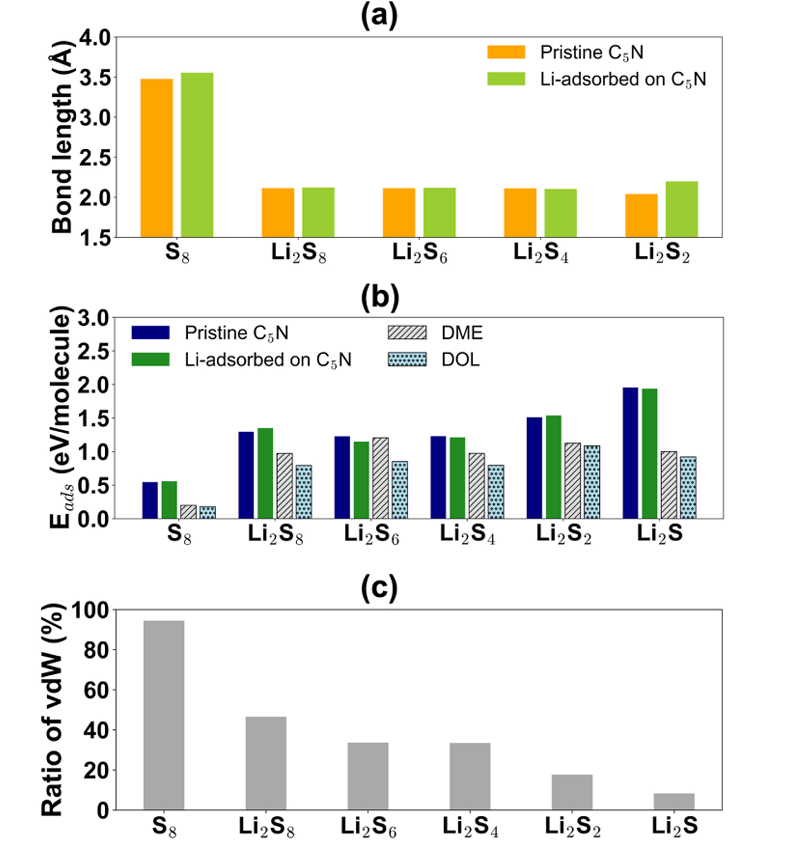
从电子结构调控的角度,理论计算阐明了C₅N促进电荷传输的微观机制。态密度(DOS)和能带对齐分析显示,C₅N的半导体带隙(1.41 eV)及功函数(5.23 eV)与碳基导电材料(如石墨烯)和常用集流体(Ni、Al等)匹配良好,有效降低了界面电荷传输阻力。
此外,多硫化物吸附引起C₅N费米能级上移和导带态密度增加,进一步增强了电子传导能力,这一发现为设计兼具强吸附性和高导电性的阴极材料提供了理论依据。
对于最终放电产物Li₂S的分解过程,理论计算同样提供了重要见解。通过爬坡弹性带(CI-NEB)方法计算发现,C₅N可将Li₂S的分解能垒降低至0.50 eV,远低于孤立Li₂S的3.27 eV以及WS₂(2.42 eV)等材料,表明其能有效促进充电过程中Li₂S的氧化分解,缓解电极钝化问题。这一结果与COHP分析中观察到的Li-S键弱化现象(ICOHP值从-0.580 eV增至-0.565 eV)相吻合,从化学键角度揭示了催化作用的本质。
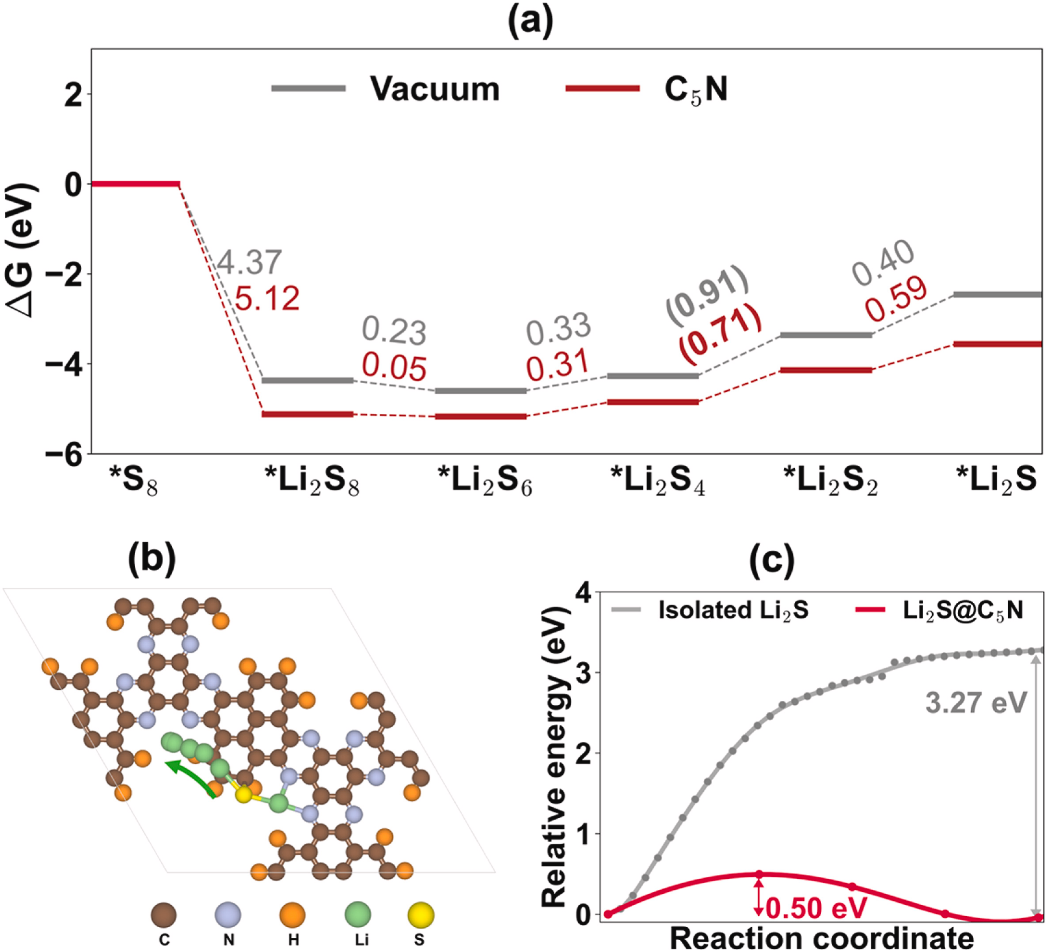
综上所述,理论计算在SRR研究中的应用,不仅从热力学和动力学角度阐明了C₅N提升LSBs性能的作用机制,还通过原子尺度的电子结构和化学键分析,建立了材料结构–吸附特性–催化活性之间的构效关系。这些研究为开发高效、稳定的锂硫电池阴极材料提供了精准的理论指导,显著缩短了实验研发周期,有力推动了LSBs从理论探索向实际应用的转化进程。