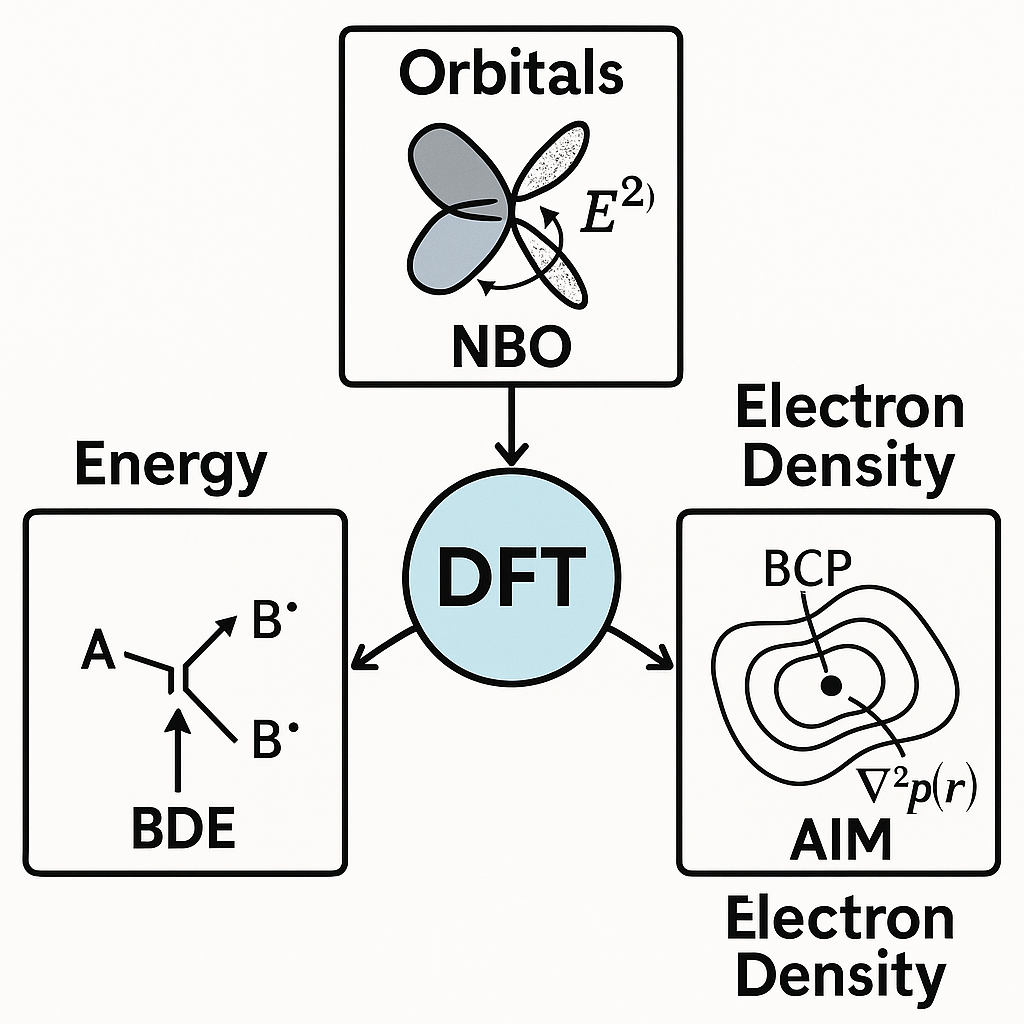
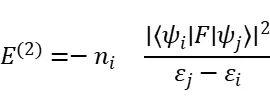本文围绕DFT框架下化学键强度分析展开。首先介绍了键解离能(BDE)方法,通过计算分子的均裂反应热力学能量差来量化键稳定性,可直观评估共价键断裂与自由基反应活化势能,但对制备能和弱相互作用存在局限。
其次阐述了自然键轨道(NBO)分析,借助局域化轨道和二阶微扰计算揭示键的电子成因与超共轭、供受电子转移等微观机制,适用于共轭体系和弱相互作用研究。
最后介绍了原子间电子密度拓扑(AIM)方法,通过识别键临界点的电子密度、Laplacian及能量密度参数来判定键本质,可对共价到非共价相互作用提供连续谱分析,但对电子密度精度要求较高。
文末总结指出,三种方法从能量、轨道与密度三维视角互补联用,能够全面解析键本质,辅助预测分子稳定性与设计新型功能材料。
化学键的强弱是分子稳定性、反应性、材料性能等一系列性质的基础,因此对其进行定量判断具有重要理论意义与实际价值。传统的经验方法如键级、键长、振动频率等虽能提供直观参考,但不足以满足复杂体系中高精度分析的需求。
随着计算化学的发展,密度泛函理论(Density Functional Theory, DFT)以其在处理中小分子体系时兼具精度与效率的优势,成为现代化学中分析化学键性质的核心工具。
在DFT计算框架下,化学键强度的分析可以从能量、轨道与电子密度三个维度进行展开。具体而言,当前主流的分析路径包括以下三种方法。
键解离能(Bond Dissociation Energy, BDE)是量化化学键强度最直接的方法之一,定义为将一个化学键均裂(homolytic cleavage)为两个自由基所需的能量。该值可以通过DFT计算体系的总能量获得。设有一个分子 AB,其均裂反应为:
其中,E(A–B)是母体分子的总电子能,E(A∙)和E(B∙)是断键后两个碎片的单点能量。所有能量都需在相同计算水平(如相同泛函和基组)下进行,包括必要时的零点能(ZPE)修正。
该方法优点在于其物理意义直观,是热力学层面上判断键强度最基础的指标。尤其在研究共价键断裂、自由基反应或材料裂解路径时具有高度应用价值。
BDE的数值越大,表示断裂该键所需能量越高,说明该键更稳定。其计算结果常用于筛选热稳定性高的分子、评估抗氧化剂的活性中心,或作为反应路径分析的能垒参数。
然而,BDE计算也存在一定局限。例如,当碎片分子具有不同电子自旋态或存在结构弛豫现象时,所计算的BDE值可能包含“准备能”(preparation energy)等非键能项,影响其本征解释。
此外,对于非共价相互作用(如氢键、π-π作用等),BDE难以准确反映键强度,需辅以其他分析方法。
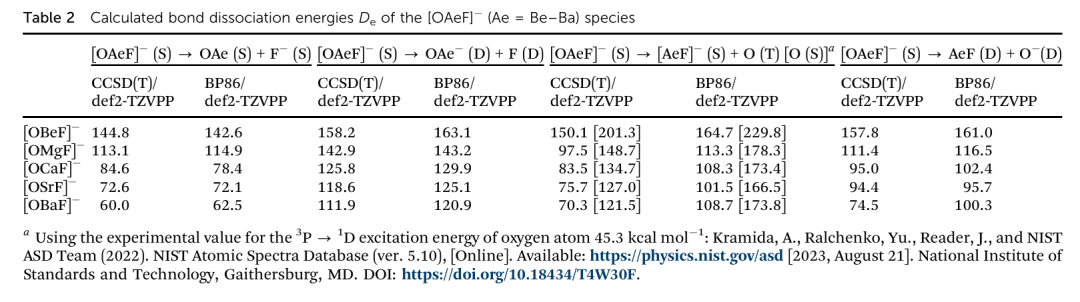
在文献 “The strongest dative bond in OAeF⁻ (Ae = Be–Ba)” 中,作者系统研究了线性阴离子 OAeF⁻(其中 Ae 为 Be、Mg、Ca、Sr、Ba)体系的几何构型与化学键性质,并提出 OBeF⁻ 中存在主族体系中最强的配位键。该结论的关键依据来自对配位键强度的键解离能(BDE)分析,其结果详见文章中的 Table 2。
doi.org/10.1039/D4CP01909A
该表列出了不同 Ae 原子构成的 OAeF⁻ 分子的 BDE(De)数值,计算方法为 CCSD(T)/def2-TZVPP 与 BP86/def2-TZVPP。结果显示:从 Be 到 Ba,BDE 值依次为 144.8、113.1、84.6、72.6 和 60.0 kcal/mol,呈现出单调递减趋势。
该趋势揭示了 Ae 原子半径增大导致其与 O、F 之间配位键稳定性降低的规律性。其中,OBeF⁻ 的 BDE 高达 144.8 kcal/mol,远高于其他体系,是目前文献中已知主族闭壳层体系中最强的配位键。
作者指出,这种极强的键强不仅源于 Be 的小尺寸和高电荷密度,还得益于三中心相互作用与轨道协同效应。通过对比 AeF⁻ 和 OAeF⁻ 两类体系的键能趋势,进一步说明了三原子配位结构中电子重分布与几何耦合在增强键强方面的关键作用。
因此,Table 2 不仅展示了不同主族金属构建的 OAeF⁻ 配位结构的热力学稳定性,更提供了一个理论上的键强上限,为未来主族元素新型配位化学设计提供了重要参考。
自然键轨道分析(Natural Bond Orbital, NBO)是一种轨道空间分析方法,旨在将复杂的分子轨道结构转化为具有化学意义的局域化轨道(如σ、π、孤对等)。NBO分析能够从微观电子结构层面揭示键的电子成因与供受机制,特别适用于分析共轭体系、超共轭作用及氢键等弱相互作用。
NBO方法的核心之一是二阶微扰分析,它通过计算电子从供体轨道(如孤对轨道 LP)向受体轨道(如反键轨道 σ*)的相互作用能 E(2),以衡量电子转移稳定化效应。公式如下:
其中,ψi为占据态供体轨道,ψj为未占据受体轨道,ni为占据数,εj、εi为轨道能量,F为Fock算符。该公式体现了轨道间电子耦合越强、能级差越小,电子转移越有利,进而键的稳定性越强。
在实际应用中,NBO可用于分析杂环分子中N–O、C=O键的超共轭稳定化,揭示氢键中供受轨道结构,以及识别金属配体键中的电荷迁移过程。与BDE相比,NBO方法不提供绝对能量指标,但可对键强来源进行解释,是理解反应机理和构效关系的重要手段。
值得注意的是,NBO分析依赖于计算程序中对轨道的分解方式,存在一定主观性。对于相似分子结构之间的对比,其相对值具有参考价值;但对于不同体系,E(2)值不可直接用于量化比较,需与其他指标联合使用。
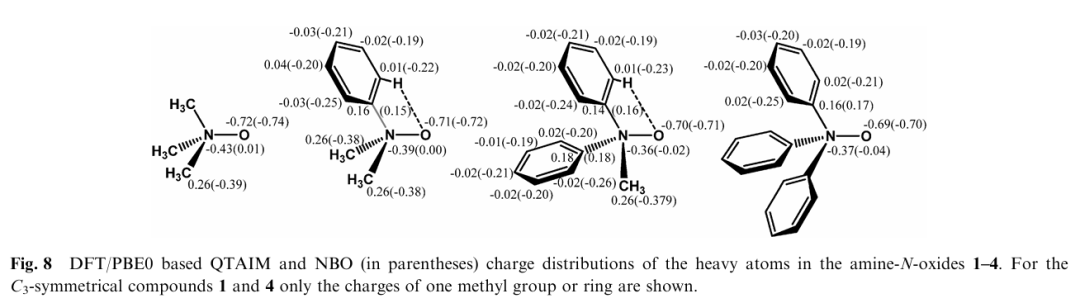
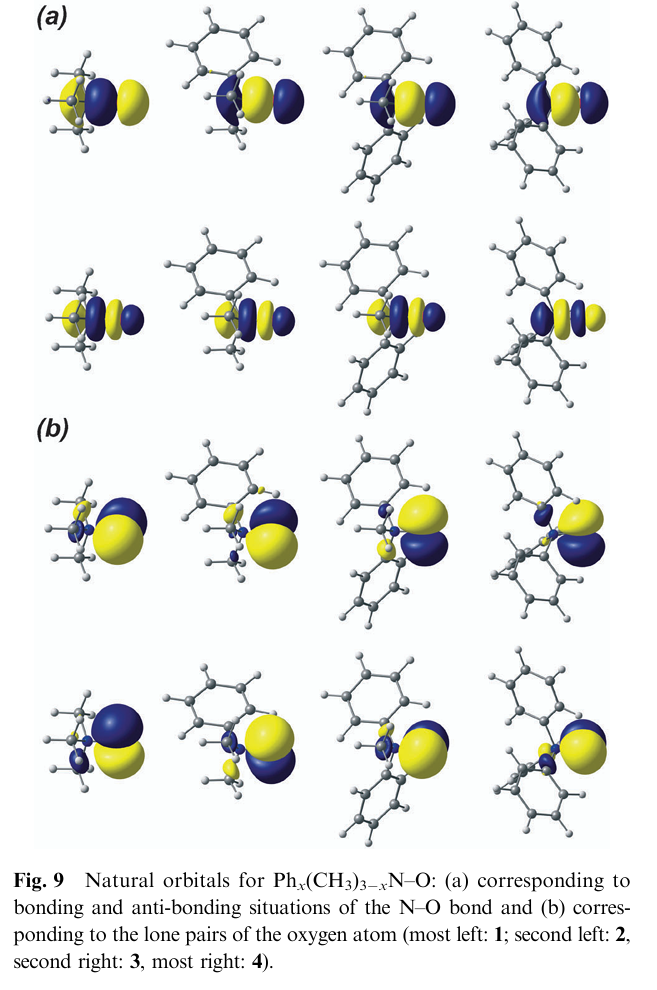
在文献 “Bonding situation and N–O bond strengths in amine-N-oxides”(Rogachev & Burger, 2012)中,NBO(自然键轨道)分析方法被用作理解N–O键电子结构及其与键强变化关系的重要手段。该方法的使用重点体现在文中NBO分析部分和图 9(Fig. 9)与图 8(Fig. 8)中。
doi.org/10.1039/C2CP22341D
作者研究了四种不同取代基的胺-N-氧化物 Phₓ(CH₃)₃₋ₓN–O(x = 0–3),虽然其N–O键的键解离能(BDE)随着苯基数量增加显著降低,但NBO分析显示,其N–O键的轨道结构在整个系列中几乎保持不变。
具体而言,图 9 展示了代表性 NBOs:N–O键主要由氮和氧的杂化轨道重叠形成,轨道组成中氮贡献约为 55%,氧贡献约为 45%,且氮的s型轨道比例显著高于氧。图 8 则比较了不同方法(NBO与AIM)获得的原子电荷分布,强调了氧原子稳定存在较大的负电荷(约 –0.7e),进一步支持其为极性共价键结构。
doi.org/10.1039/C2CP22341D
这说明:尽管BDE降低,NBO分析揭示了N–O键在各分子中的成键方式本质上无变化,因此BDE变化并非源于键本身弱化,而是来自对应胺分子的结构弛豫能(Preparation Energy)差异。
作者指出,苯基数量增加导致胺部分从平面向四面体转变所需的构型重组能增加,从而显著影响BDE。这一推论通过NBO轨道组分与电荷分析得到了支持,也反映出NBO分析在从微观轨道层面揭示化学键“强度来源”方面的重要价值。
原子间电子密度拓扑分析(Atoms in Molecules, AIM)由Bader提出,是一种基于实空间电子密度的化学键分析方法。它不同于轨道分析,而是通过研究电子密度 ρ(r)的拓扑结构来界定和判断原子间的化学键。
AIM的核心在于识别分子中的键临界点(Bond Critical Point, BCP),即两个原子之间电子密度梯度为零、电子密度鞍点的位置。通过分析该点的电子密度值 ρBCP、Laplacian ∇2ρBCP以及能量密度参数(如动能密度G、势能密度V),可以判断键的本质与强度。
闭壳层相互作用(如氢键、离子键):ρBCPa.u.,∇2ρ>0
此外,对于氢键等非共价作用,Espinosa 等人提出可用势能密度 V(r) 估算其键能,公式为:
AIM分析具有极强的可视化能力,可通过拓扑图、电子密度图等形式呈现键的存在与强度。在多重键、多中心键、非典型弱相互作用(如CH···π、X···Halogen键)中尤为重要,成为结构化学中的标准工具之一。
不过,AIM方法依赖于高质量电子密度分布,因此对计算精度和基组要求较高。此外,对于含有过渡态、强电子相关或金属簇类体系,AIM解释需要与其他方法联合使用,以避免误判。
在文献 “DFT, AIM, AND NBO Analyses of 1-Methyl-2-thioxoimidazolidin-4-one Tautomers and Their Complexes with Iodine” 中,AIM(原子间电子密度分析)方法被用于从电子密度拓扑的角度探讨 MTIO 分子及其碘配合物中的 S–I 相互作用强度,具体分析结果详见文中 Table 8。
doi.org/10.1134/S0022476612040063
该表列出了所有配合物的S–I 键关键点(BCP)处的电子密度 ρ(r)、Laplacian ∇²ρ(r)、能量密度和相关距离参数。
研究发现:在所有复合物中,平面构型的配合物(如 T1–I₂ 和 T4–I₂)在 S–I BCP 处的电子密度值最高(ρ ≈ 0.025–0.026 a.u.),且 S–I 键长最短(约 3.055–3.065 Å),表明这些配合物具有更强的相互作用。这些关键点的 Laplacian 为正(∇²ρ > 0),能量密度较小,表明 S–I 键具有闭壳层(非共价)性质。
通过对比不同构型和异构体的 AIM 数据,作者指出,键强的增强主要归因于 C=S 双键的共轭增强作用,使得硫原子更具电子供体能力,从而与碘分子形成更有效的轨道重叠。
因此,AIM 分析不仅确认了 S–I 弱配位键的性质,也通过电子密度分布清晰地揭示了结构构型对键强的调控机制,为新型碘吸附剂的构建提供了理论支撑。
综上所述,密度泛函理论(DFT)为化学键强度的量化与解析提供了多维度、系统化的理论工具。本文所述三种方法——键解离能(BDE)、自然键轨道(NBO)分析、以及原子间电子密度拓扑分析(AIM)——各自从能量、轨道、电子密度三个角度出发,为研究者提供了互补的分析视角。
BDE方法以能量差值为核心指标,适合评估共价键、自由基反应等过程中键的稳定性,其结果具备明确的热力学意义。
NBO方法则从分子轨道角度解释键的电子成因,能够揭示供受电子转移、超共轭效应等微观机制,是解析非经典键结构与反应路径的重要工具。
AIM方法基于实空间电子密度,能够描述从共价键到非共价相互作用的连续谱,特别适合于识别氢键、配位键和弱相互作用等复杂电子结构。
三者在实际研究中往往不是孤立使用,而是协同分析。例如,BDE可提供宏观键强的量化依据,NBO和AIM则能从微观结构与密度分布上提供解释支撑。
通过这种多维度方法联用,研究者可更全面地理解键的本质、预测分子稳定性,甚至设计具有特定键性质的新型材料与功能分子。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!