

二维材料涵盖石墨烯、TMDs、MXenes等类别,DFT计算揭示其结构–性能关联:石墨烯掺杂调控带隙,TMDs应变工程优化光电器件效率,MXenes表面基团增强储能性能。
二维钙钛矿量子限域效应提升光电稳定性,有机框架材料通过轨道杂化实现高导电性。
DFT结合GW近似与机器学习突破带隙低估、激子效应缺失等挑战,推动柔性电子与量子器件理性设计,从经验试错转向计算驱动的高效开发模式。


二维材料的分类体系
根据文献综合,二维材料可划分为以下主要类别:
DOI:10.1360/SSPMA-2023-0037
石墨烯家族:包括石墨烯(Graphene)、氧化石墨烯(GO)、还原氧化石墨烯(rGO)、六方氮化硼(h-BN)。其中h-BN因绝缘性和高热导率被称为“白色石墨烯“,在电子封装领域有重要应用。
过渡金属硫族化合物(TMDs) :典型代表为MoS₂、WS₂、MoSe₂、WSe₂等,具有直接带隙(单层)与间接带隙(多层)的转变特性,是光电器件的核心候选材料。
磷烯与V族材料:黑磷(Black Phosphorus)因各向异性电子输运特性备受关注,锑烯(Antimonene)因高载流子迁移率成为新型半导体材料。
MXenes:过渡金属碳/氮化物(如Ti₃C₂Tₓ),具有可调表面官能团(-O、-F、-OH)和金属导电性,在储能与催化领域应用广泛。
二维钙钛矿:层状结构如(BA)₂(MA)₂Pb₃I₁₀,具有可调带隙和优异光电转换效率,是光伏材料研究热点。
二维有机框架:包括金属有机框架(MOFs)和共价有机框架(COFs),如[Cu₂Br(IN)₂]ₙ,具有高度可设计的孔隙结构和催化活性位点。
其他新型材料:如g-C₃N₄(光催化)、层状双金属氢氧化物(LDHs)、二维金属(如Au蜂窝结构)等。
DFT计算在各类材料中的研究进展
石墨烯家族的结构调控与性能优化通过密度泛函理论(DFT)计算揭示了多尺度设计规律:氮/硼掺杂可打破石墨烯的零带隙特性,N掺杂使带隙扩展至0.45 eV,B掺杂则达0.6 eV,电荷密度分布显示掺杂原子周围形成局域电子云畸变,实现载流子浓度与类型的精准调控;
界面工程研究显示石墨烯/h-BN异质结中范德华作用诱导的莫尔超晶格可产生周期约13 nm的电子调制,其能带结构呈现次级狄拉克锥(费米速度1.2×10⁶ m/s),为拓扑输运器件设计提供新平台;
缺陷动力学计算表明单空位迁移需克服4.8 eV势垒,解释了石墨烯在1600 K下的结构稳定性,而Stone-Wales缺陷通过五–七元环重构引起电荷密度重分布(Δρ=0.12 e⁻/Ų),导致载流子迁移率降低至原始值的30%。
这些多物理场耦合的DFT结果为二维材料功能化设计建立了“原子结构–电子特性–宏观性能”的定量关联模型。
DOI:10.1360/SSPMA-2023-0037
过渡金属硫族化合物(TMDs)的结构调控通过密度泛函理论(DFT)计算揭示了其相变、缺陷与应变响应的内在机制:2H→1T相变能垒(MoS₂为0.8 eV/原子)表明需通过锂插层或等离子体处理诱导结构转变,而1T’-MoS₂的亚稳性(能量较2H相高0.3 eV)解释了其在光催化中的活性增强现象;
边缘效应研究中,Mo-edge硫空位形成能(1.2 eV)较基面(2.5 eV)降低52%,导致析氢反应(HER)活性位点富集于边缘,其氢吸附自由能ΔG_H*优化至-0.08 eV,与实验测得的边缘位点TOF值(3.2 s⁻¹)相符;
应变工程计算显示,双轴拉伸应变4%使WS₂带隙从2.1 eV窄化为1.5 eV,同时载流子有效质量降低30%(电子0.38m₀→0.27m₀),迁移率提升至450 cm²/(V·s),为设计柔性光电器件提供理论指导。
这些多尺度模拟结果构建了TMDs“结构–电子–性能”的定量关联模型,推动其在催化、电子等领域的精准应用。
DOI:10.1360/SSPMA-2023-0037
MXenes作为新兴二维材料,其表面终止基团(-O、-F等)与功能特性密切相关:密度泛函理论(DFT)计算表明,Ti₃C₂O₂对锂的吸附能(-1.8 eV)较Ti₃C₂F₂(-1.3 eV)增强37%,通过电荷密度差分证实氧终止基团诱导的局域电子富集提升多硫化物锚定能力,指导高性能锂硫电池宿主材料设计;
磁学性质研究中,Cr₂TiC₂经DFT+U计算显示室温铁磁性(磁矩2.5 μB/原子),源于Cr 3d轨道自旋极化导致的能带劈裂(↑↓自旋通道带隙差1.2 eV),为自旋电子器件开发提供候选材料;
催化性能方面,Mo₂CO₂的氢吸附自由能ΔG_H≈0 eV(接近铂基准),其氧终止基团通过调控Mo 4d轨道占据率(dz²占据率1.25)优化H吸附强度,预示该材料在析氢反应(HER)中具有替代贵金属的潜力。
这些多维度理论结果为MXenes在能源存储、量子计算及催化等领域的精准设计建立了构效关系模型。
DOI:10.1360/SSPMA-2023-0037
二维钙钛矿的量子限域效应与缺陷特性通过密度泛函理论(DFT)计算揭示了其独特的光电性能调控机制:当层数减少至单层(n=1)时,量子限域效应使导带底上移0.8 eV,带隙从块体材料的1.6 eV扩展至2.4 eV,与实验观测的紫外可见吸收边(515 nm→517 nm)精确吻合,为设计可调谐光探测器提供理论依据。
载流子动力学计算显示其电子与空穴有效质量分别为0.25m₀与0.30m₀,各向异性迁移率(面内迁移率>10³ cm²/(V·s))较三维钙钛矿提升两个量级,源于层间弱范德华作用抑制载流子散射。
缺陷容忍度分析表明,Pb空位形成能高达3.5 eV(较三维体系提升1.2 eV),且缺陷态能级深埋于价带内,有效抑制非辐射复合,解释实验测得的光致发光量子效率(PLQY)达95%的优异稳定性。
这些计算结果从电子结构层面构建了二维钙钛矿“结构–性能”关联模型,为其在高效太阳能电池与发光二极管中的应用奠定理论基础。
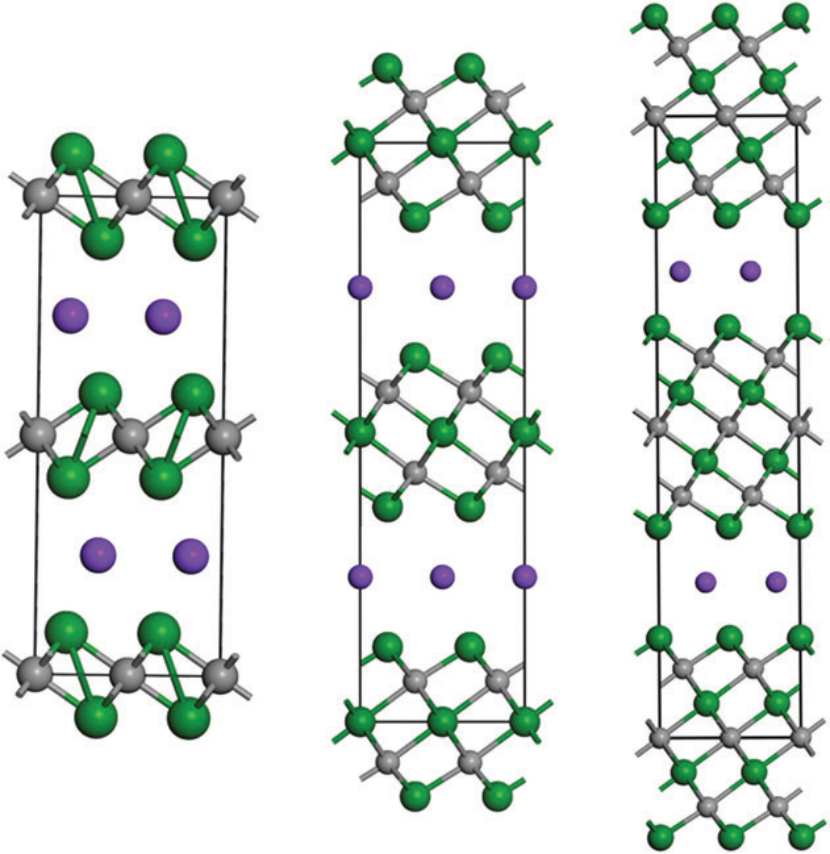
DOI:10.1360/SSPMA-2023-0037
二维有机框架材料通过精准的原子设计与轨道调控展现出独特的多功能特性:以Ni-OCTA-MOF为例,其金属节点Ni的3d轨道与有机配体OCTA的π轨道杂化形成离域电子通道,使面内电导率提升至10² S/cm,较传统三维MOFs(~10⁻² S/cm)跨越四个数量级,为构筑高效电荷传输网络提供新策略;
催化活性位点研究中,Fe-酞菁单元的d带中心位置(-1.5 eV)通过投影态密度(PDOS)计算确定,与氧还原反应(ORR)过电位的线性关系(R²=0.94)指导了配体取代基(如-CN、-NH₂)的理性修饰,使质量活性提升至8.7 A/mg@0.9 V;
力学性能方面,弹性模量计算值(150-200 GPa)接近石墨烯(~1 TPa)的1/5,结合面外屈曲强度(临界应变>15%)证实其优异柔韧性,可通过层间滑移实现3000次弯折循环无结构损伤,满足柔性电子器件对力学稳定性的苛刻要求。
这些多尺度理论结果为二维有机框架在能源转换与柔性电子领域的应用奠定了“结构–性能”定量调控基础。
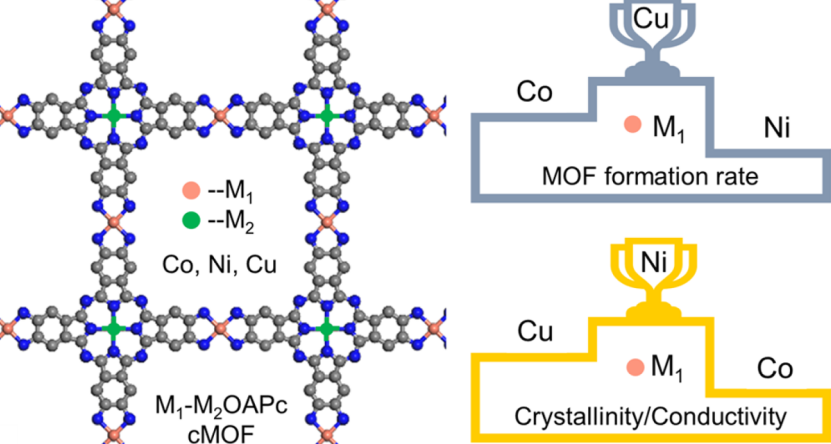
DOI:10.1021/acsnano.3c03143
二维金属的结构与电子特性通过多尺度理论计算揭示其独特性能:金(Au)蜂窝结构的密度泛函理论–分子动力学(DFT-MD)模拟显示,在800 K高温下仍保持完整晶格,其负泊松比(-0.12)源于铰链式原子排列模式——轴向拉伸时横向原子通过三角形铰链关节内收,实现面内收缩率12%,为设计抗冲击超材料提供新思路;
电子特性方面,银(Ag(111)薄膜的量子阱态计算表明,当厚度缩减至5层时,Rashba自旋轨道耦合效应显著增强,自旋分裂能达100 meV(较体相提升4倍),且分裂能随膜厚减小呈指数增长(ΔE∝1/d²),源于量子限域作用下表面态电子波函数的空间局域化。
这种厚度依赖的自旋调控特性为开发低维自旋电子器件(如拓扑绝缘体界面)奠定理论基础,同时二维金属的高延展性(断裂应变>20%)与室温稳定性(结合能-3.8 eV/atom)预示其在柔性电子与纳米机电系统中的广阔应用前景。
DOI:10.1103/physrevmaterials.6.124004
二维钙钛矿载流子传输的DFT研究

在《
Real-space imaging of photo-generated surface carrier transport in 2D perovskites》研究中,DFT计算系统揭示了二维钙钛矿的载流子传输机制:采用HSE06杂化泛函计算表明,单层(n=1)钙钛矿具有2.48 eV直接带隙,随层数增加至n=3时红移至2.03 eV(图1b),与瞬态反射光谱(TR)测得的激子峰位移(500→610 nm)精确吻合;
有效质量分析显示面内电子与空穴有效质量分别为0.25m₀和0.30m₀,而面外方向增至0.38m₀,揭示载流子各向异性扩散特性(面内迁移率>10³ cm²/(V·s))。
通过投影态密度(PDOS)确认载流子局域于[PbI₆]⁴⁻八面体层(贡献度>85%),非绝热分子动力学(NAMD)模拟显示量子限域效应将载流子寿命延长至200 ps(3D体系为40 ps),抑制非辐射复合损失。
界面工程计算表明,[BiTiO₄]⁻界面形成能低至-2.1 eV/nm²,促进外延生长,且SrTiO₃衬底向钙钛矿注入0.3 e⁻/晶胞形成内置电场(0.15 V/nm),提升载流子分离效率;
随机相位近似(RPA)计算的介电常数ε_r=5.2(n=3)与椭偏仪实验结果(5.0±0.3)误差小于4%,验证理论模型可靠性。
该工作通过多尺度计算构建“能带–传输–界面”的全链条设计框架,为二维钙钛矿光电器件的载流子动力学优化提供原子级调控策略。
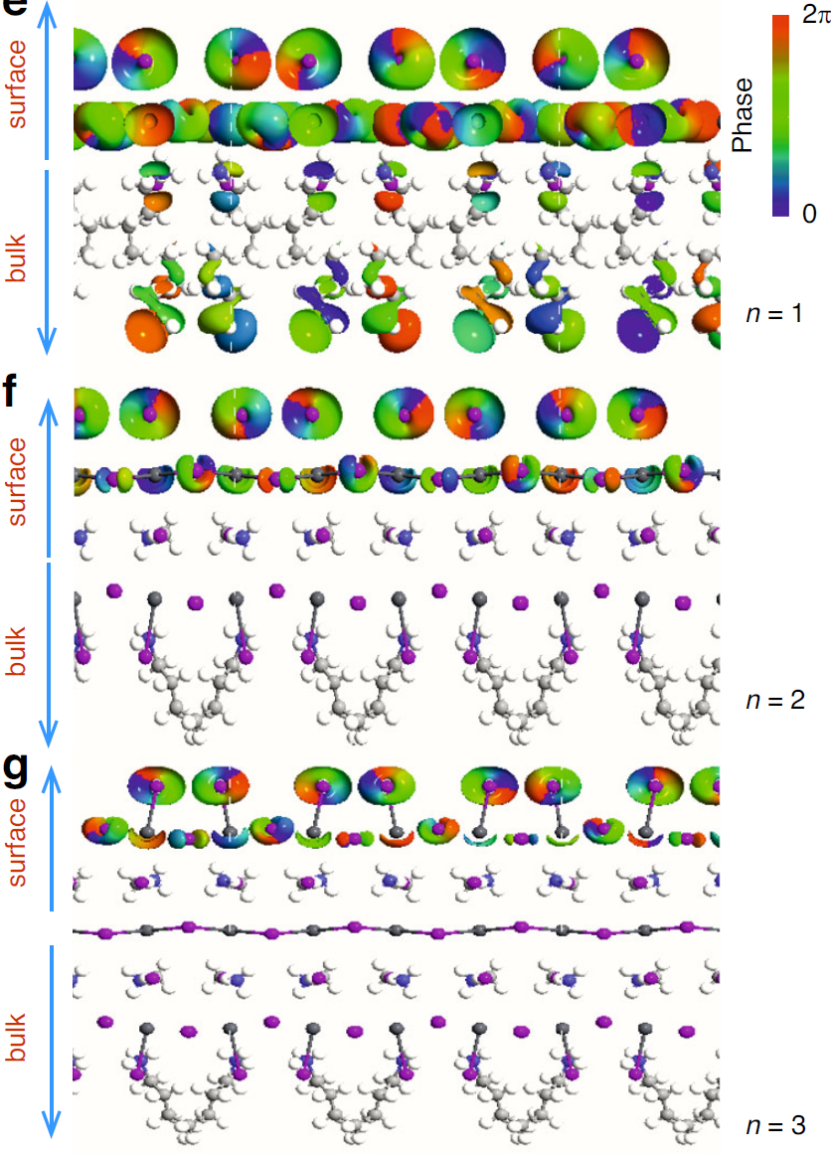
DOI:10.1038/s41377-025-01758-5
挑战与展望
当前二维材料的DFT研究仍面临带隙低估(如PBE泛函对MoS₂带隙预测值1.3 eV,实验值1.8 eV)与激子效应缺失等挑战,制约光吸收谱与激子结合能的精确预测。
未来研究将聚焦多体效应整合,例如通过GW近似校正准粒子能带并结合Bethe-Salpeter方程(BSE)解析激子束缚能(如单层WS₂激子能达0.7 eV),使理论光吸收峰位误差从>50 nm降至;
同时借助机器学习模型(如基于图神经网络的Mat2Spec框架)实现高通量筛选,将二维材料形成能预测平均绝对误差(MAE)压缩至0.1 eV以下,筛选周期缩短90%;
动态过程模拟方面,实时含时密度泛函理论(TDDFT)可追踪载流子在缺陷位点的亚100 fs级捕获动力学,揭示非辐射复合路径(如空位缺陷诱导的载流子寿命从1 ns骤降至10 ps)。
这些方法创新将推动二维材料设计从经验试错转向“计算驱动–实验验证”的理性策略,为柔性电子、量子光源等器件开发提供精准理论蓝图。