

Q1:朱老师,我这种环状结构有机物,我是应该选择其中某一个碳原子吸附到吸附位点上去,还是选择这个环的中心吸附到吸附位点上去呢?
A:参照文献 选择,或者都尝试,选择能量最低的
Q2:朱老师,Mn的赝势用标准的赝势还是用Mn_pv呢?我看vaspkit默认是pv
A:都试试看差异,或者vaspkit默认
Q3:朱老师,我计算乙醇在表面脱氢过程,中间过程涉及到的分子都要先优化后在进行吸附吗?
A:可以构建吸附模型直接优化
Q4:朱老师,请问如图所示,如果我想算一下某个多元环有机物吸附在晶体(NaYF4)表面的性质,我应该如何确定初始模型呢?是拿有机分子的对称中心(比如图上的三元环中间的hallow)去对晶体表面的各个可能的吸附点位(Na, Y, F, bridge)等等吗?这样的话,可能的结构太多了,有没有更简单的判断方法,或者我就随意放置,它可能优化到一个比较合适的结构吗?
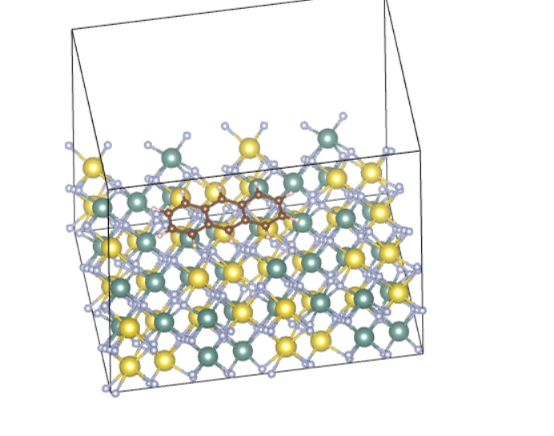

A:严格的做法是尝试不同位点优化,选择能量最低的那个
Q5:朱老师,请问在一个催化剂有多个晶面时,例如(001)(010)(011)(110),选择切哪一个晶面的标准有哪些呢(我师姐跟我说看别人文献里的,但我感觉有点笼统,没有选择的依据)
A:参考文献中的选择,或者选择XRD低指数晶面
Q6:朱老师,有没有说计算这几个晶面的某个性质,然后筛选下的
A:切面算表面能,然后选择表面能最小的表面
Q7:朱老师,在水相中计算吸附能,和平时计算吸附能,有什么区别吗?INCAR中需要改动哪些参数呢?
A:主要是要加水分子在模型中
Q8:朱老师 我用这个结构去算吸附能是不是太大了横向距离大约10埃以上
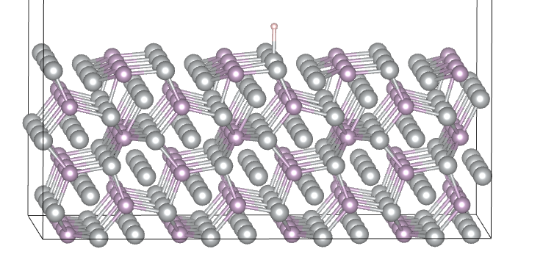
A:10-15埃
Q9:朱老师,在画差分电荷图的时候,vesta中的isosurface的“positive”指的是电子聚积的意思吗?
A:是的
Q10:老师,我想问一下他这个Co就是按照比例随机掺杂的吗?
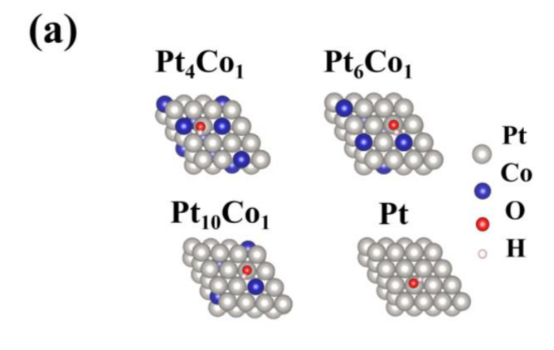
A:具体看文章中说明吧,有可能随便选,有可能均匀分布
Q11:朱老师,请问吸附算查分电荷密度的话,是将吸附后的结构进行优化,然后算一下整体的电荷密度。然后在整体的POSCAR中分别直接删去B部分和A部分的原子,然后对剩下的部分算电荷密度ρA和ρB,再将整体的电荷密度减去ρA和ρB,对吗?
A:是的