

然后,详解X 射线吸收光谱学吸收边的产生、能量变化,阈值能量与结合能在不同体系中的情况,以及 XAFS 在孤立原子、分子与凝聚态体系中的表现和 XANES 与 EXAFS 的区分;
最后深入解读 EXAFS,包括其唯象模型、函数表达式,以及从 EXAFS 光谱中可获取的平均原子间距、配位数、局域无序程度等信息,同时提及分析难点与现有技术支持。想学习更多知识,请参考以往内容。
1895 年 11 月,威廉・伦琴发现 X 射线,其高穿透性、对原子序数和密度的依赖性等特性,使其在数月内便应用于医学放射造影。
1912-1913 年,首批 X 射线衍射实验完成,同时莫塞莱研究了 X 射线波长与玻尔原子模型能级能量的关系,自此 X 射线成为研究物质微观性质的关键工具。
X 射线吸收系数的精细结构(XAFS)早在 1920 年被发现,但受限于技术,长期无法有效利用。
20 世纪 70 年代,同步辐射源的应用及解读方案的发展,让人们能通过 XAFS 获取物质局域几何与电子结构的定量信息。R. 施图姆・冯・博德韦尔曾撰写 EXAFS 早期发展的详细历史。
X 射线束的通量 Φ 指单位时间内穿过光束单位横截面的光子数量。当 X 射线束穿过厚度为 x、横截面积大于光束横截面积的样品时(图 1.1,左图),通量会按照指数规律衰减,其中 μ 是样品的线性衰减系数。
线性衰减系数 μ 取决于 X 射线的能量以及样品的成分和密度。单一原子种类的贡献以质量吸收系数 μ/ρ 的形式被制成表格:
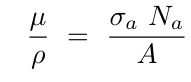

其中ρ 是密度,σₐ是原子衰减截面,Nₐ是阿伏伽德罗常数,A 是原子质量。
X 射线束的衰减主要源于三种不同机制(图1,右图)
1️⃣在光子能量相对较低时,主导相互作用是光电吸收:光束中的一个光子因与原子相互作用而被吸收;
光子能量被原子吸收,原子可能被激发(一个或多个电子被激发到更高的束缚能级),也可能发生电离(一个或多个电子被发射出去,后一种现象称为光致电离)。
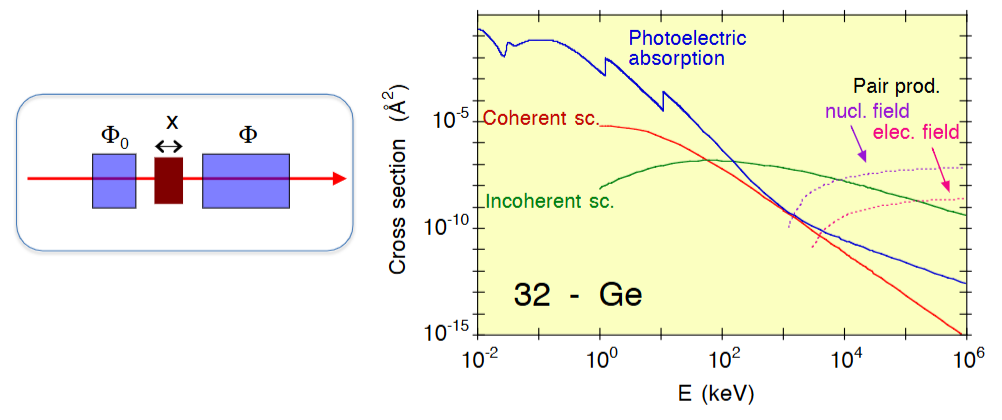
图1: 左图:一束 X 射线穿过厚度为的样品。右图:锗中 X 射线衰减不同机制的原子截面:实线 = 光电吸收,虚线 = 非修饰散射(汤姆逊散射),点线 = 修饰散射(康普顿散射),点划线 = 对产生。
2️⃣第二种相互作用是散射,在低能情况下,其作用弱于光电吸收。光子与原子碰撞后会偏离原来的运动轨迹。单个光子与单个电子的散射可分为两种情况:
(1)修正散射(康普顿散射):散射过程导致光子波长发生改变,波长的正向变化量为Δλ=λc (1-cosθ),其中 θ 是散射角,λc =0.002426Å 是康普顿波长。根据量子力学,自由电子对光子的散射始终是修正散射。
(2)非修正散射:散射过程中光子波长不发生改变,即Δλ=λ’-λ=0,因此能量 E’=E。非修正散射对应于经典汤姆森理论中自由电子对 X 射线的散射。
对于束缚在原子中的电子,修正散射和非修正散射的统计平衡,取决于发生康普顿散射的光子损失的能量ΔEcom≈ħωinΔλ/λin 是否大于电子的结合能。
当考虑 X 射线在原子集合体中的散射时,可分为以下两类:
(1)弹性散射:光子能量守恒,即ħωout=ħωin。
(2)非弹性散射:光子能量不守恒。非弹性散射不仅可能源于单电子层面的康普顿效应,还可能源于光子与电子集体激发或振动集体激发的相互作用。
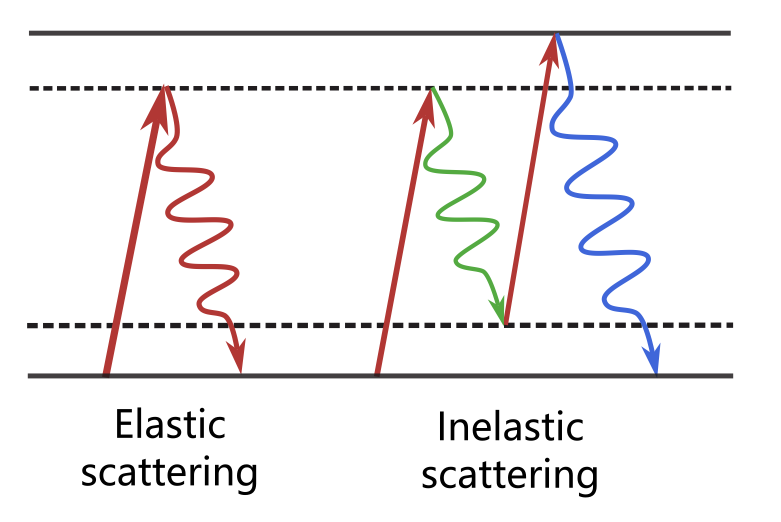
图2:弹性散射与非弹性散射
(图源:http://www.ahos.com.cn/index/info/7783)
3️⃣在高能情况下,主导相互作用是电子对产生:光子湮灭,产生一对正负电子(e⁺e⁻)。该过程要求光子能量大于约 1MeV(两倍电子静质量能量),且无法在真空中发生。为保证线动量守恒,光子必须与外部场(核场或电子场)相互作用。
在 1MeV 以下的能量范围内,存在光电吸收和散射两种相互作用,由此产生了三类基本的实验技术:
(1)光谱学基于吸收机制,主要用于获取物质的电子结构信息。根据研究对象是 X 射线吸收随能量的变化还是 X 射线发射随能量的变化,可分为 X 射线吸收光谱学和 X 射线发射光谱学。在光电子光谱学中,会收集并分析样品吸收 X 射线光子后发射出的电子。
(2)弹性散射可用于获取凝聚态体系(晶体和非晶体固体以及液体)的原子尺度几何结构信息;晶体固体的弹性散射通常被称为衍射。非弹性散射可用于获取原子中电子动量分布(通过康普顿散射)以及原子集体激发的相关信息。
(3)成像技术基于样品不同部位对 X 射线的衰减程度差异,可获得样品的宏观图像,包括医学和工业放射造影以及 X 射线显微术。吸收和散射都会对成像所依赖的衰减产生影响。
本文重点关注能量范围在约 1keV 至约 100keV 之间的 X 射线,其对应的波长分别约为 0.12Å 至 12Å。(能量ħω(单位:keV)与波长 λ(单位:埃)之间的关系为ħω=12.398/λ)。
在这个能量范围内,主导的衰减机制是光电吸收。因此,我们可以将线性吸收系数与线性衰减系数μ(ω) 视为等同。
此外,在该能量范围内,光电吸收系数的变化规律非常简单,且不同原子种类的光电吸收系数在定性上极为相似。通过双对数图像(如图1右图和图3左图),能最清晰地展现这些特性。
随着被吸收 X 射线光子能量的增加,吸收系数 μ(ω) 逐渐减小(能量越高的 X 射线穿透能力越强);吸收系数也与原子种类相关(原子量越大的原子,吸收能力越强)。光电吸收系数的平滑能量依赖性会被尖锐的突变—— 吸收边所中断。
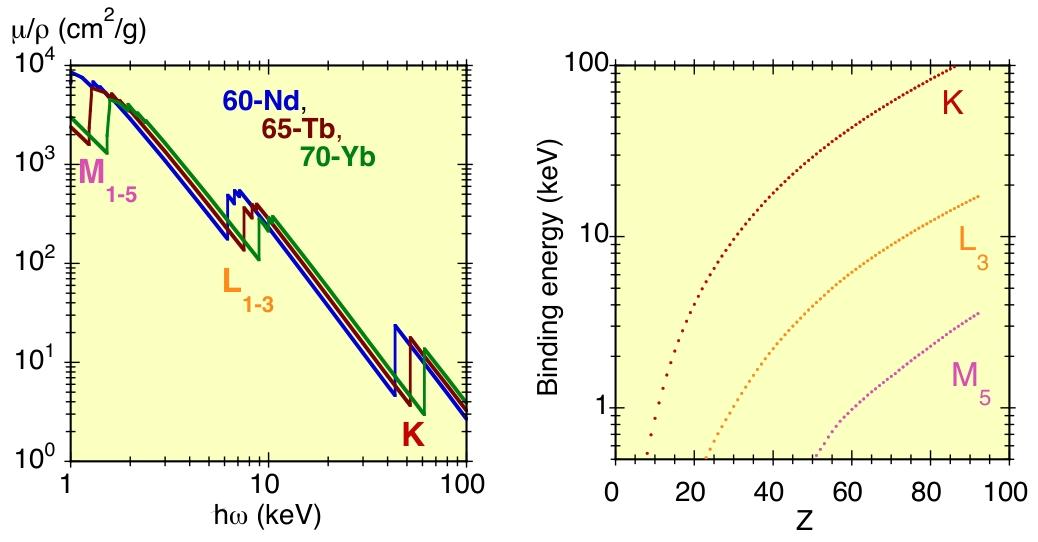
图3:左图为钕(Nd)、铽(Tb)、镱(Yb)在 1-100 千电子伏特(keV)能量范围内的 X 射线质量吸收系数;右图为 K、L₃、M₅芯能级中电子的结合能随原子序数(Z)的变化关系。
吸收边(图1右图、图3左图)的产生,是由于当光子能量足够高,能够将电子从较深能级激发出来时形成的。
出于历史原因,X 射线吸收边用连续的字母 K、L、M…… 来标记,它们分别对应主量子数 1、2、3……(图 3)。能量最高的吸收边是 K 边,对应电子从最深的 1s 能级被激发出来的过程。
接下来能量较高的是三个 L 边:L₁边对应电子从 2s 能级被激发,L₂边和 L₃边对应电子从 2p 能级被激发。
L₂边和 L₃边的区别源于原子轨道角动量与自旋角动量的不同取向,这种不同取向会导致能量产生不同的自旋 – 轨道贡献,其相关信息汇总于表 1.1(表中还包含了初始 d 壳层填满的原子的 M 边信息)。
表 1.1:高能吸收边(按光电子能量递增顺序排列)及涉及的电子壳层构型

如图3 右图所示,各类吸收边的能量随原子序数 Z 的增加而单调递增。例如,K 边能量从氢原子的 13.6eV(远低于 X 射线区域)增加到铀原子的 115.606keV。
在 X 射线能量区域(1-100keV),两个相邻吸收边之间的吸收系数与光子能量和原子序数的关系近似为:
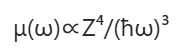
吸收边能量通常定义为特定核心能级电离的阈值,因此它与核心能级的结合能相对应。由于结合能随原子序数 Z 单调递增(图3,右图),每个吸收边能量都对应着一种特定的原子种类。这种特性正是 X 射线光谱技术具有原子种类选择性的根源。
✅孤立原子
吸收 X 射线光子后,根据 X 射线能量的不同,孤立原子可能出现以下两种情况:
(1)激发:若光子能量低于结合能,且与核心能级和较高的未占据束缚能级之间的能量差相等,那么核心能级的电子会被转移到未占据的束缚能级。
(2)电离:若光子能量高于结合能,电子(即光电子)会从原子中被发射出来。
电子能级的结合能是将电子从原子中移出所需的最小能量(电离能)。
与光学区域的吸收系数相比,X 射线吸收系数的变化规律更为简单,且不同原子种类的 X 射线吸收系数具有相似性,这是因为核心能级的结合能远大于未占据束缚原子能级之间的能量差。
X 射线吸收光谱主要由核心能级激发主导,受外层能级结构的影响很小。然而,外层能级的分布会导致吸收系数在吸收边附近出现精细结构,这使得精确确定阈值能量变得并非更为复杂。
✅分子与凝聚态体系
对于分子和凝聚态体系,光致电离过程比孤立原子更为复杂。在分子中,光电子在逸出前可能与周围原子发生复杂的相互作用,因此确定阈值能量比原子体系更困难。
在凝聚态体系中,光电子通常无法逸出(除非在表面附近发生光致电离,这种情况会产生光电子发射光谱)。对于金属,结合能定义为核心能级与费米能级之间的能量差;对于半导体,结合能定义为核心能级与价带顶之间的能量差。
✅结合能表格
目前已有的表格中,气体的电子结合能以真空能级为参考(电离能),金属的电子结合能以费米能级为参考,半导体的电子结合能以价带顶为参考。
表 1.2 列出了部分元素的 L 边和 K 边的结合能。L 边之间的间距随原子序数 Z 的增加而增大。具体而言,L₂边和 L₃边的能量差源于自旋 – 轨道效应,且该差值随原子序数 Z 的增加而增大(图4)。
表 1.2:部分元素 K 边和 L 边的结合能(单位:eV)
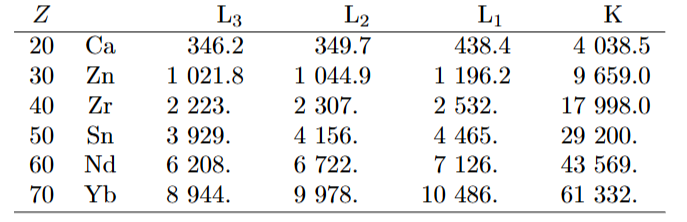
图3(左图)中吸收系数的平滑变化只是一种近似情况。实际上,在吸收边附近,吸收系数会呈现出精细结构,即所谓的 X 射线吸收精细结构(XAFS)。
✅孤立原子
对于孤立原子(惰性气体、金属蒸气),XAFS 仅存在于吸收边附近几 eV 的能量范围内,它反映了核心电子向未占据束缚能级(里德伯能级)的跃迁。
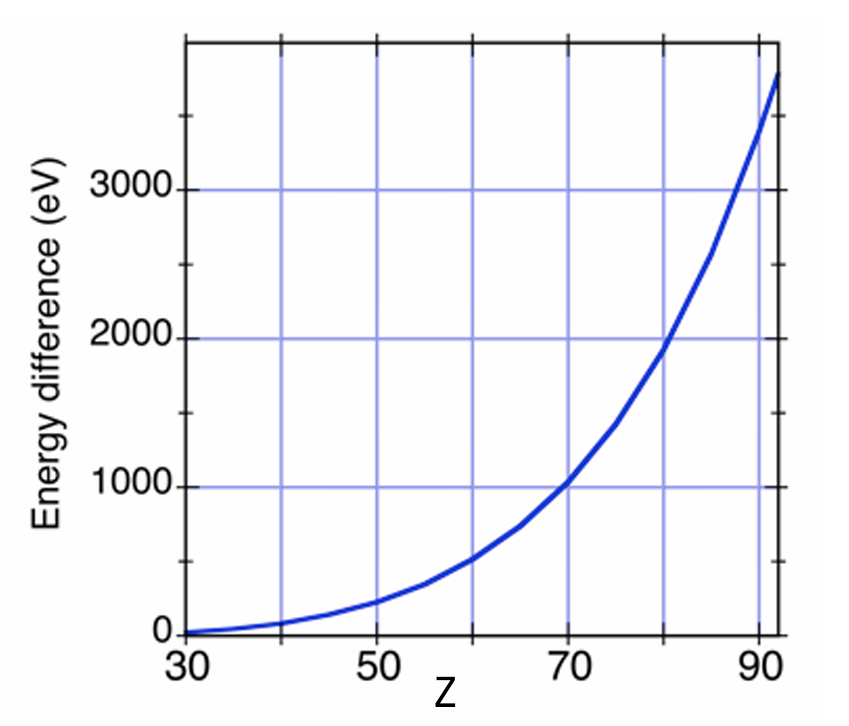
图4:L₂和 L₃边的自旋 – 轨道分裂随原子序数Z的变化关系。
图5左图展示了氩原子 K 边附近极窄能量范围内的情况。向里德伯能级的跃迁会产生洛伦兹形结构(图中的虚线峰),其宽度由激发态核心态的能量不确定性决定,约为 1eV,远大于光学跃迁的典型宽度。
电离阈值对应里德伯系列的上限;阈值的阶梯状变化由于激发态核心态宽度的影响,转变为反正切曲线。通过将理论计算与实验光谱进行最佳拟合,确定了 Eb=3205.9eV 的值。
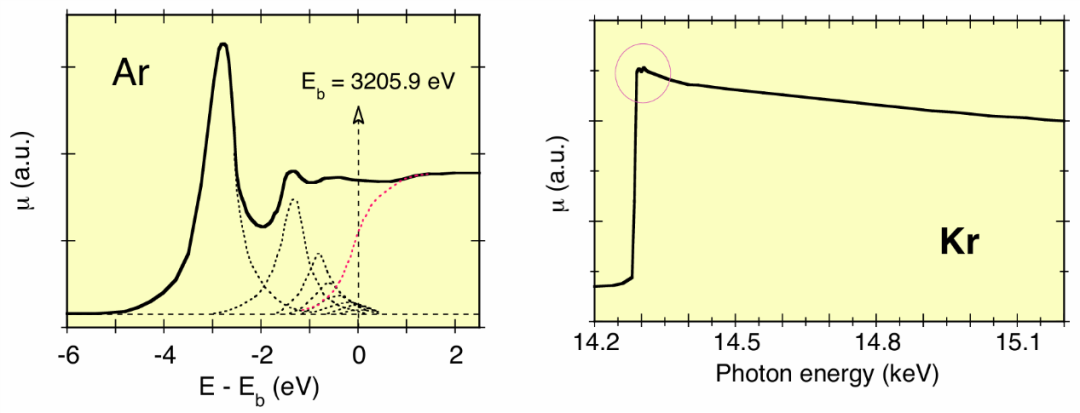
图5:氩(左侧,连续线)和氪(右侧)K 边的精细结构,其中氩的结果。
图5右图展示了氪原子 K 边以上较宽能量范围内的情况。由于激发态核心态宽度随原子序数的增加而增大(且大于氩原子的激发态核心态宽度),向里德伯能级跃迁产生的精细结构仅表现为吸收边处的小峰(圆圈内部分)。
在吸收边以上的能量区域,吸收系数的变化如预期的孤立原子那样,呈现出完美的平滑性。
✅分子与凝聚态体系
在分子气体和凝聚态体系中,XAFS 会受到吸收原子周围其他原子的强烈影响,其范围可延伸至吸收边以上 1000eV 甚至更高的能量区域。
图6左图展示了分子 N₂的 K 边情况。
除了在 405-410eV 范围内向原子里德伯能级的跃迁(其振幅已放大 10 倍,这些跃迁汇聚于电离阈值)外,还有其他现象对 XAFS 产生贡献,例如向振动激发态的跃迁(400-402eV 范围内的边前结构)、双激发(415eV 附近)以及所谓的形状共振(约 420eV 处的凸起)。
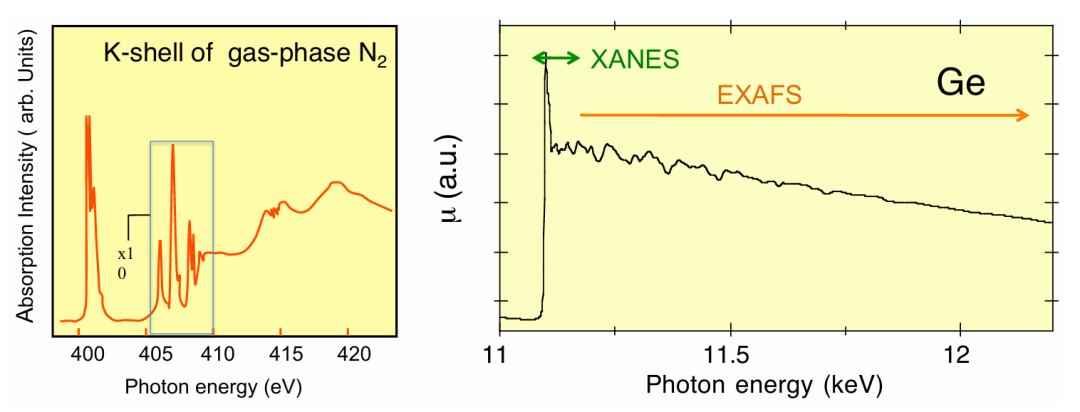
图6右图展示了晶体锗的 K 边情况。此处的精细结构在吸收边以上至少 1000eV 的能量范围内都能观察到。
✅命名法
XAFS 通常分为两个区域:
吸收边以上 30-50eV 范围内的结构称为 XANES(X 射线吸收近边结构)或 NEXAFS(近边 X 射线吸收精细结构)。
通过 XANES,可以获取物质的局域电子结构和几何结构信息。有时,吸收边附近几 eV 范围内的特征会被区分为边前结构和边结构。
从 XANES 区域延伸至通常约 1000eV 能量范围的精细结构称为 EXAFS(扩展 X 射线吸收精细结构)。
EXAFS 包含特定原子种类周围的局域几何结构信息。目前,EXAFS 的解读方法已相当成熟,且比 XANES 的解读更为简单。
实验测得的 X 射线吸收系数包含了不同电子壳层激发的贡献。以图 3 左图为例,当光子能量为 20keV 时,2s、2p、3s、3p、3d…… 能级的电子可被激发,但 1s 能级的电子无法被激发;当光子能量为 80keV 时,1s 能级的电子也能被激发。
为了有效解读 XAFS,通常会考虑仅一个核心能级对吸收系数的贡献,例如 1s 能级(图 5)。在 K 边能量以下,1s 电子的贡献为零。
通常,特定吸收边处吸收系数及其精细结构的基本特征可通过单电子过程来解读,但多电子激发也可能产生不可忽视的贡献。
X 射线的光电吸收可通过混合方法进行合理解释:原子采用量子力学描述,而电磁场采用经典近似描述。原子与电磁场的相互作用在含时微扰理论的框架内进行处理,一级近似下可得到所谓的 “费米黄金定则”。
根据费米黄金定则,吸收系数与相互作用哈密顿量的矩阵元的平方成正比,该矩阵元连接了相互作用前原子的定态(初始态)和相互作用后原子的定态(最终态)。
由于初始态是原子的基态,与光子能量无关,因此吸收系数的精细结构只能归因于最终态。下面我们将定性地分析最终态随光子能量的变化情况,为简化分析,仅考虑单电子跃迁。
✅孤立原子
对于孤立原子,当光子能量低于光致电离能时,最终单电子态是离散的里德伯能级。只有当光子能量与从核心能级到里德伯能级的允许跃迁能量相等时,吸收系数才不为零。
当光子能量高于光致电离能时,最终态是自由电子态的连续谱,吸收系数没有精细结构。因此,研究孤立原子的 X 射线吸收系数实用价值不大。
✅分子与凝聚态体系
当吸收原子嵌入分子或凝聚态体系中时,XAFS 的研究具有更为重要的意义。此时,吸收边以下的离散光谱会因新的最终态可能性而变得更加丰富,例如分子的振动特性、半导体中的激子能级等。
即使光子能量高于光致电离能,吸收原子周围的环境也会对最终态产生影响。因此,XAFS 能够反映局域环境引起的电子结构变化,而局域环境又与局域几何结构相关。
在吸收边以上约 20-30eV 的能量范围内,计算光电子的最终态通常并非易事。有两种主要的、原则上等效的方法可供选择:一种基于分子轨道理论,另一种基于电子多重散射理论。这两种方法都能建立 XAFS 与局域结构之间的联系。
散射方法最显著的优势在于,当光电子能量高于约 20-30eV 时,单散射在决定最终态方面起主导作用,多重散射通常可以忽略不计。
单散射的处理相对简单,这使得我们能够利用仅包含少数结构参数的解析表达式来解读 XAFS。
因此,吸收光谱中 XANES 和 EXAFS 两个区域的区分,可以通过不同的解读方法来解释。
在 EXAFS 区域,可以采用相对简单的单散射形式主义来解读 XAFS。而在 XANES 区域,解读 XAFS 需要考虑多重散射效应,有时也可采用分子轨道方法。
通常会采用一种简单的唯象模型,在单散射方法的框架下解释 EXAFS 的起源(图7)。当能量足够高的 X 射线光子被原子吸收时,一个核心电子会从原子中被发射出来(电离过程)。
光电子的动能等于光子能量与核心结合能的差值。出射的光电子可用波函数描述,在图7 中近似为球面波,其波长随光子能量的增加而减小。
如果吸收原子不是孤立的,光电子可能会被相邻原子反向散射,形成入射球面波。出射波和入射波之间的相位关系取决于光电子的波长和原子间距。因此,最终的光电子定态是出射球面波和入射反向散射波的叠加。
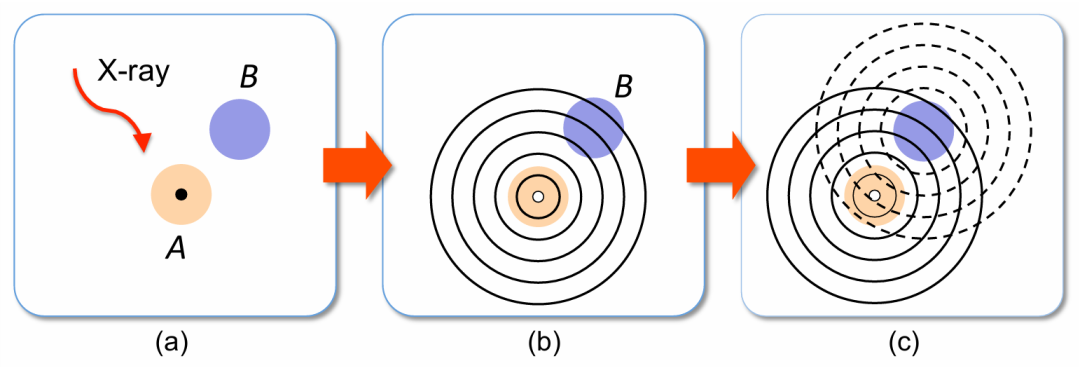
图7:EXAFS 现象的示意图解:原子 A 吸收 X 射线光子(左),光电子波的发射(中),光电子被原子 B 背散射(右)。
相位关系随光子能量的变化会影响核心位置处最终态的振幅,从而产生干涉现象,使吸收系数发生调制。EXAFS 振荡的频率取决于吸收原子与反向散射原子之间的距离,其振幅与反向散射原子的数量成正比。
作为一种结构探测手段,EXAFS 具有两个主要特点:
1. 原子种类选择性:通过将 X 射线能量调至相应的吸收边来实现。
2. 对长程有序不敏感:这一方面是由于光电子波的球面特性,另一方面是由于光电子的平均自由程较短,通常限制在约 10Å 范围内。
EXAFS 振荡通常用归一化的无量纲函数表示:
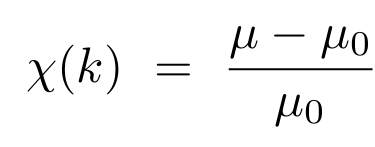
图8(右图)给出了一个 EXAFS 函数的示例。在式(1.4)中:
a) k 是光电子的波数,定义为 k=√[(2m/ħ²)(ħω-E_b)],其中ħω 是光子能量,Eb 是核心电子的结合能;
b) μ 是测得的吸收系数(图 8左图中的红线);
c) μ₀是孤立原子的吸收系数(图8 左图中的蓝线)。
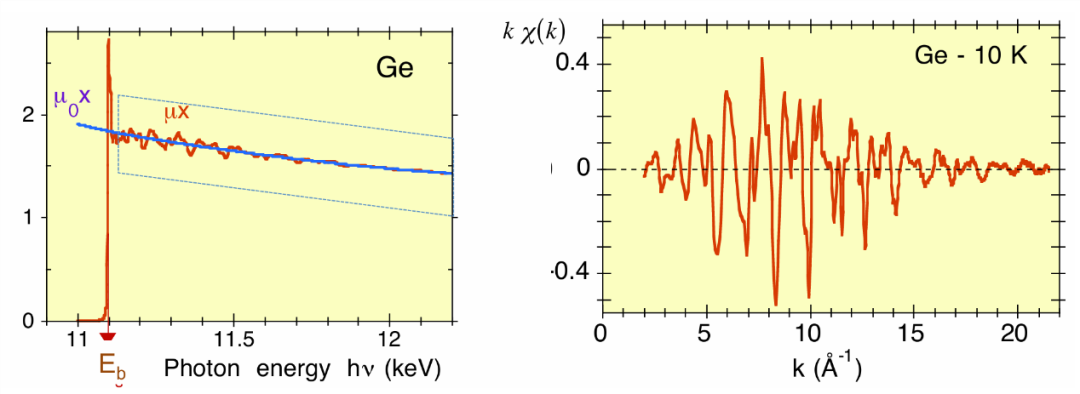
图8:左图:锗的 K 边处的实验吸收系数μx,在T=10K下测量,以任意单位绘制为光子能量hν=ℏω的函数。右图:对应的加权 EXAFS 函数Kχ(K),绘制为光电子波数K的函数。
通过分析 EXAFS 光谱,可以获取吸收原子周围几个配位壳层(通常在约 10Å 范围内)的基本信息。图 8(右图)所示的 EXAFS 函数是多个配位壳层贡献的叠加。
每个配位壳层的贡献都是类似正弦曲线的函数,如图9 所示。原则上,对于每个配位壳层,至少可以获取三个重要参数:
1. 平均原子间距:与 EXAFS 振荡的频率成正比(图 9左图)。
2. 配位数 N:与 EXAFS 信号的振幅成正比(图 9中图)。
3. 局域无序程度:包括热无序和结构无序,用德拜 – 沃勒因子 exp (-2kσ²) 来衡量,它会导致 EXAFS 信号衰减(图9右图)。
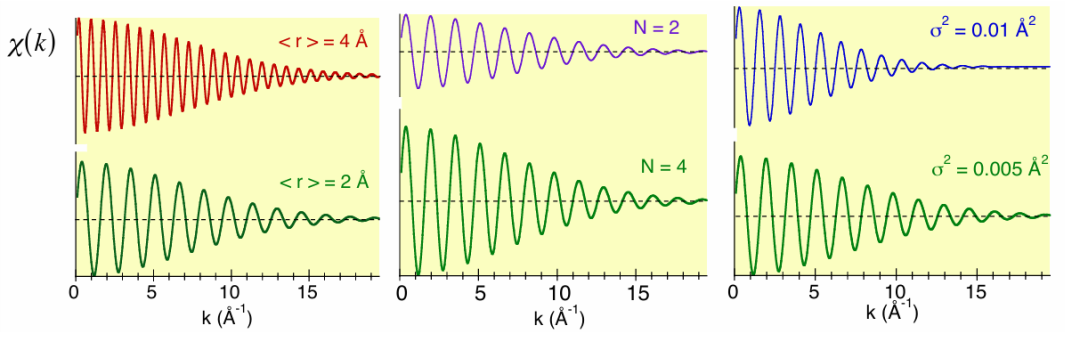
图9:给定配位壳层的 EXAFS 基本信息:频率与原子间距离成正比(左),振幅与配位数成正比(中),衰减取决于热无序和结构无序的程度(右)。
要从实验光谱中获取上述参数(、N 和 σ²)并非易事。需要妥善考虑光电子与发射原子和反向散射原子的相互作用,以及不可忽视的多体效应。
从 EXAFS 光谱中获取信息的数量和质量,取决于实验数据的质量以及可用振荡的数量(即信号的长度)。为了进行精确的定量分析,信号应至少延伸到 k≈15Å⁻¹,这对应于光子能量至少延伸到吸收边以上 900eV。
原则上,对于在 K 边测量的 EXAFS,只要样品中不存在其他元素的吸收边限制可用光谱的上限,都能满足上述要求。但对于 L 边,情况则有所不同。
在 LⅢ 边测量的 EXAFS 可能会受到 LⅡ 边的严重限制;在 L 边测量的 EXAFS 可能会受到 LⅡ 边残留 EXAFS 的干扰。对于轻元素,由于其 L 边的能量非常接近,这些限制尤为明显;而对于重元素,这些限制会逐渐减弱(见表 1.2)。
【高端测试 找华算】
华算科技是专业的科研解决方案服务商,精于高端测试。拥有10余年球差电镜拍摄经验与同步辐射三代光源全球机时,500+博士/博士后团队护航,保质保量!
?已助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果在Nature&Science正刊及子刊、Angew、AFM、JACS等顶级期刊发表!
?立即预约,抢占发表先机!