

Q1:朱老师,在计算CO2RR过渡态的时候,如果以H作为质子供体,那初态是要把氢和CO2都吸附在催化剂表面吗?整条路径中需要加多个氢,那这些氢是在一开始就都放在催化剂表面,还是一步只放一个?放H的位置有什么要求吗,单原子的话放在基底上,结构优化一下就行么?
A:过渡态放一个就可以,位置靠近吸附物
Q2:朱老师,我之前构建的是纯金属111面和二氧化钛的异质结,我在做实验的时候,加入了另一种金属,然后发现他们分相了,这是我查文献看见的双金属相的结构构造,所以我就按照这个结构,将之前的纯金属111面中的其中一列的原子替换成了我加入的金属像我发的这张图一样,然后在与二氧化钛构建了异质结,这是可行的吗?
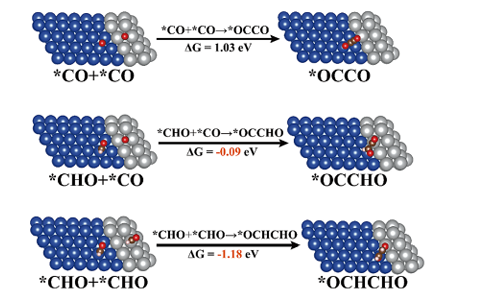

A:可以,相当于横向的异质结,保证下失配率不要高于10%
Q3:朱老师,计算功函数时,使用脚本vtot处理后,VLINE文件中第一列表示什么呢 第二列是能量吧 ?我看功函数都是用分数坐标作为横坐标 这个能转换为分数坐标吗?
A:第一列是z轴取点数,比如100,就是在z轴上均匀取100个点,最后一个点正好是z的最大值
Q4:朱老师,如何读出静电势的数值呢,就像费米能级一样,能得到具体数值?
A:有locpot文件,里面有电势分布
Q5:朱老师,请问CO2RR算到CH4结束,第一个台阶和最后一个台阶的能量差是多少呀?我翻了一些文献,没有找到给出能量差的?
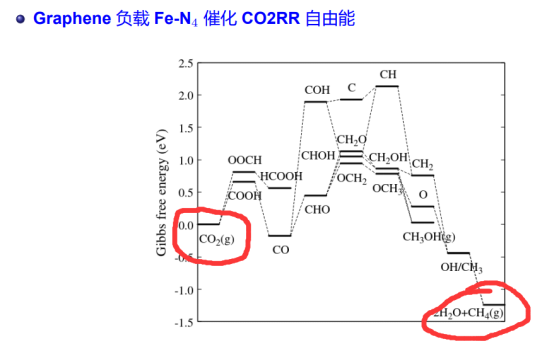
A:查热力表就可以,几个气体的自由能差别,我当时自己算的,是1.3多,以查表为准
Q6:朱老师,请问计算NO3-到*NO3的时候会进行矫正吗,因为NO3-是带电子的
A:no3–的计算方法可以参照课上her中oh–的方法
Q7:朱老师,氮化碳体系200个原子体系,算态密度都正常 算能带结构算不动(内存不足) 可能是什么原因? 设置还是能带所需的计算资源就是更多?
A:可能是k点太多了
Q8:朱老师,请问我算完差分电荷密度后,在VESTA中作图顶端出现了这个蓝色的圆点,这是计算有误吗?
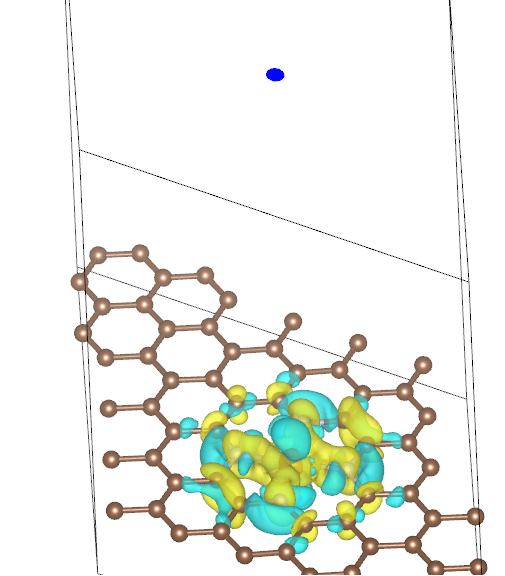
A:不是计算错误,是由于周期性的原因,可以把c方向的周期显示的小一些
Q9:朱老师,这是氮化碳材料的布里渊区对称点,能带计算时路径应该怎么取? 高对称点是延z轴的好像没有意义?
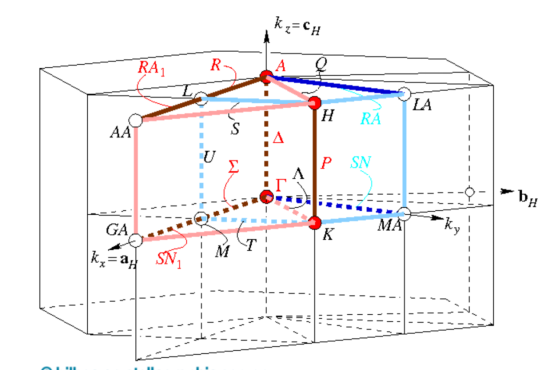
A:对于体相结构,也可以到materials project数据库中,搜一下这个结构,里面一般会给出能带信息以及高对称点的选择
Q10:朱老师,想请教一下,我算结构优化的时候 3h就完成了 但是我跑DOS,电子步很慢,我该怎么调整
A:加一个 istart=1和icharge=11
本次答疑由拥有15年VASP实战经验的华算科技朱老师(同济大学本博、深圳海外高层次人才)提供。?点击进入《VASP | 理论计算常见问题解答专题》,快速查找更多计算解决方案。