

说明:本文华算科技介绍了离子液体的基本概念、分类、性质及其在化学、能源和环境领域的应用,重点阐述了密度泛函理论(DFT)、分子动力学(MD)和从头算分子动力学(AIMD)等计算方法在解析离子液体微观机制中的作用。
离子液体(Ionic Liquids, ILs)是一类由有机阳离子和无机或有机阴离子组成的、熔点低于100°C的液态盐类。与传统溶剂相比,离子液体具有低挥发性、高热稳定性、宽液体范围和可调的化学性质等优势,广泛应用于催化、分离、能源储存和电化学等领域。

DOI: 10.1021/acs.chemrev.5b00763
根据化学组成和性质,离子液体可分为以下几类:
室温离子液体(RTILs):在室温或接近室温下呈液态,通常由大体积、不对称的阳离子(如咪唑鎓、吡啶鎓或季铵盐)和阴离子(如[BF4]⁻、[PF6]⁻或[NTf2]⁻)组成。
功能化离子液体:通过引入特定功能基团(如羟基、氨基或氟化烷基),实现特定化学或物理功能,如提高溶解性或催化活性。
双亲性离子液体:兼具亲水和疏水特性,适用于两相体系的分离或乳化。
生物基离子液体:由可再生资源(如氨基酸或糖类)衍生,绿色环保,适合可持续应用。
离子液体的核心特性是其可调性,通过改变阳离子和阴离子的组合,可精确调控其物化性质,如密度、粘度、导电性和溶解能力。例如,[BMIM][BF4]因低粘度和高导电性常用于电化学器件,而[NTf2]⁻阴离子的疏水性使其适合液–液萃取。
计算方法为揭示离子液体的微观结构和动态行为提供了核心工具。以下为三种主要计算方法:
1、密度泛函理论(DFT)
DOI: 10.1016/j.cej.2024.154287
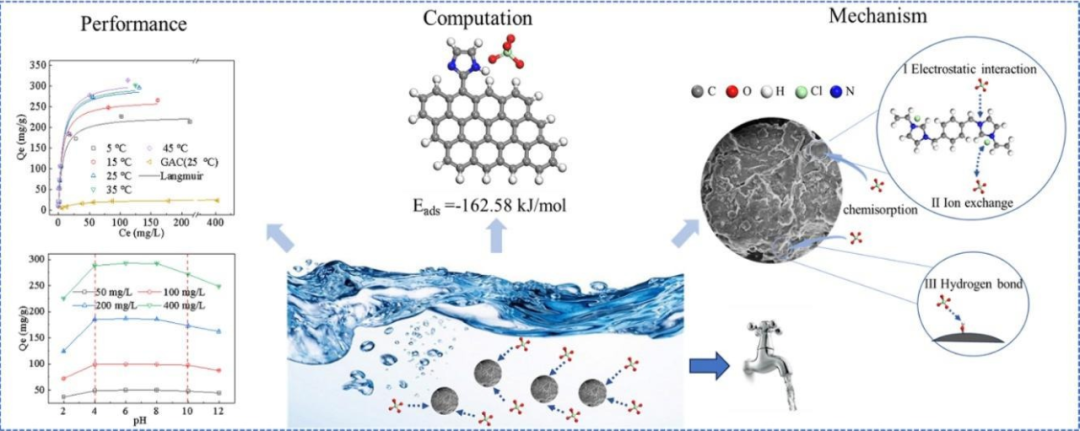
密度泛函理论通过计算离子液体的电子结构,揭示离子间的电荷分布、键合特性及相互作用机制。它可预测溶解能力、反应活性及热力学性质。
例如, DFT计算表明,高氯酸盐对咪唑 N 具有更好的吸附能(−162.58 kJ/mol),突出了其对其他 N 和 O 官能团的潜在选择性。PIL-GAC 在 25 °C 时的最大吸附容量(Qm)为 300.05 mg/g,比原始 GAC 高 12 倍,优于以前报道的吸附剂。此外,DFT能分析电荷转移和轨道杂化,为离子液体在催化反应中的活性提供理论依据。
2、分子动力学(MD)
DOI: 10.1016/j.cej.2021.129168
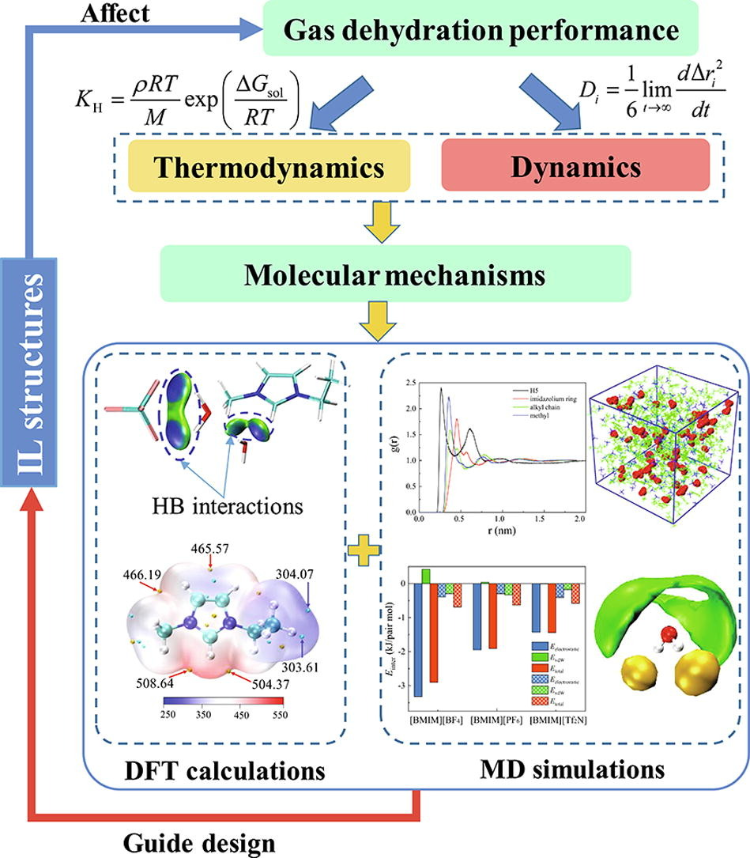
分子动力学基于经典力学,模拟离子液体中离子和分子的动态行为,揭示扩散系数、径向分布函数(RDF)和分子取向等特性。MD能捕获温度、压力和溶剂对离子液体结构的影响。
例如,通过QC计算和MD模拟揭示了分子水平的微观机理,结果表明,同时具有最小阳离子和阴离子尺寸的IL(即[EMIM][BF4])同时对应于最强的氢键(HB)H2O-阴离子和最强的HB相互作用以及H2O-阳离子的范德华相互作用。
3、从头算分子动力学(AIMD)
DOI: 10.1021/acscatal.5c01833
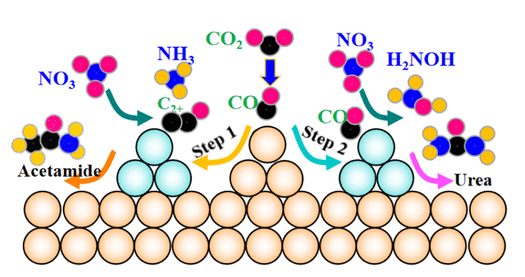
从头算分子动力学结合量子力学和经典动力学,实时模拟离子液体中电子和原子的动态行为,特别适合研究离子液体在复杂环境中的反应性。AIMD能揭示离子液体与溶质分子的动态相互作用,如分子解离或界面重构的过程。
例如,采用从头算分子动力学(AIMD)方法,通过研究实际CO2RR条件下O-H和C=O键长变化的动态结构演化,研究溶剂层对反应中间体的影响。结果表明,在300 K的[Bmim][BF4]条件下,在pH依赖性微环境中,CuPd NPs在模拟时间10 ps后保持稳定。[Bmim][BF4]的pH依赖性微环境在设计C-N和C-C偶联的高性能催化剂中起着重要作用。
离子液体作为一类独特的液态盐类,凭借其低挥发性、可调性和高稳定性,在化学、能源和环境领域具有广泛应用前景。密度泛函理论、分子动力学和从头算分子动力学等计算方法通过解析电子结构、动态行为和反应路径,深化了离子液体微观机制的理解。
这些研究不仅推动了离子液体设计的优化,还为绿色化学、能源储存和环境治理等领域的技术创新提供了支持。未来,计算方法的进步将进一步提升离子液体的性能和应用范围。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???