VASP(Vienna Ab initio Simulation Package)是一款基于密度泛函理论(DFT)的量子力学模拟软件,广泛应用于材料科学、化学、物理学等领域。
其核心计算方法之一是自洽场(Self-Consistent Field, SCF)算法,解决Kohn-Sham方程,该算法通过迭代计算电子波函数和密度,直到系统达到自洽状态。
自洽计算是VASP计算中最基础也是最重要的步骤,其目的是找到电子波函数和相应的总能量,使得电子在晶胞中的分布达到自洽。自洽计算通常包括电荷密度的初始化、波函数的迭代优化以及能量和力的计算。
VASP计算需要准备超算连接软件EASYCONNECT与SSH,建模软件VESTA,超算连接软件Winscp
jp-minerals.org/vesta/en/download.html
EasyConnect下载-EasyConnect最新版下载V7.6.7.0
Downloading WinSCP-6.5.3-Setup.exe :: WinSCP
ISTART=0 #开始新的任务,随机产生初始波函数
ICHARG=2 #开始新的任务,从原子电荷密度产生体系初始电荷密度
EDIFF=1E-5 #相邻两步电子迭代的能量差收敛标准
EDIFFG=-0.1 #离子弛豫的force的收敛标准
ISMEAR=0 #费米能级附近电子占据数为高斯分布,适合金属、半导体、绝缘体
Automatic generation #注释行
修改 INCAR 文件,让VASP执行自洽计算,修改IBRION=-1、NSW=0、LAECHG=.T.
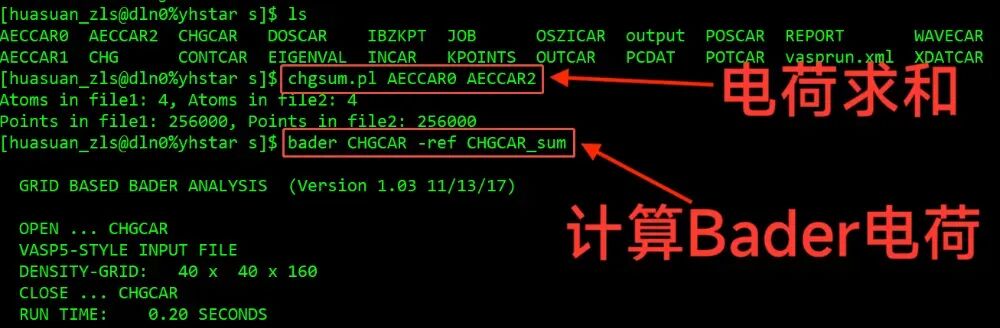
第四步,chgsum.pl和bader脚本处理数据
chgsum.pl AECCAR0 AECCAR2
bader CHGCAR -ref CHGCAR_sum
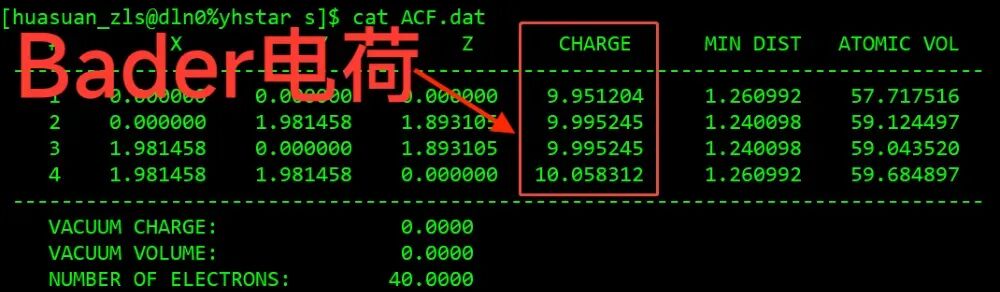
Bader电荷结果在ACF.dat文件中的CHARGE一列
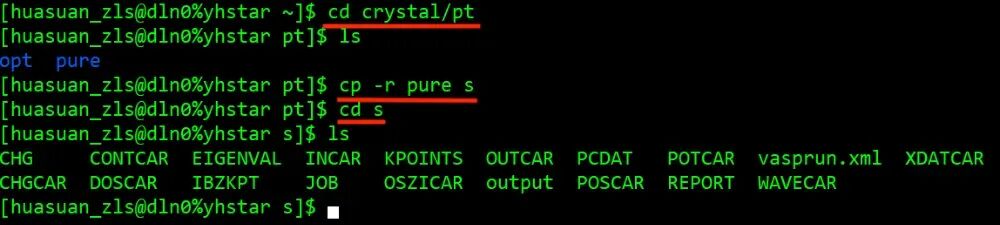
华算科技朱老师给大家介绍 pt001 的Bader电荷计算。Bader电荷是要依托于自洽计算完成的,首先之前已经做好了 001 面的结构优化计算,在pure 文件夹中,那么这里做一次自洽计算,把 pure 复制成 s, 进到自洽在计算文件夹 s 里面,第一步把CONTCAR变成POSCAR结构保留下来,然后修改INCAR。先进行自洽参计算的参数修改, IBRION =-1, NSW =0,然后加上Bader电荷的标签, LAECHG =.T. ,这个会输出原子、芯电子和价电子的一些信息,后续用脚本可以得到Bader电荷,也就是每个原子上的电荷数值,它并没有曲线图,它是以列表的形式展示的。
自洽计算算完之后可以看到得到了 AECCR0 文件和 AECCAR2文件,还需要用到 CHGCAR 这样三个文件进行 Bader 的分析。第一步,使用脚本 chgsum.pl,这是求和的一个脚本。把这个AECCR0和AECCAR2文件的电荷密度相加完之后,它会输出一个CHGCAR_sum文件,然后再使用 Bader 命令 bader CHGCAR -ref CHGCAR_sum 把这两个文件做一次处理,得到Bader电荷数据。最后的结果输出在 ACF. dat 文件中,可以看一下这里的 CHARGE一列,就是每个pt原子上的Bader电荷,那么要分析它的电子转移的话,可以和赝势中的价电子进行对比。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!