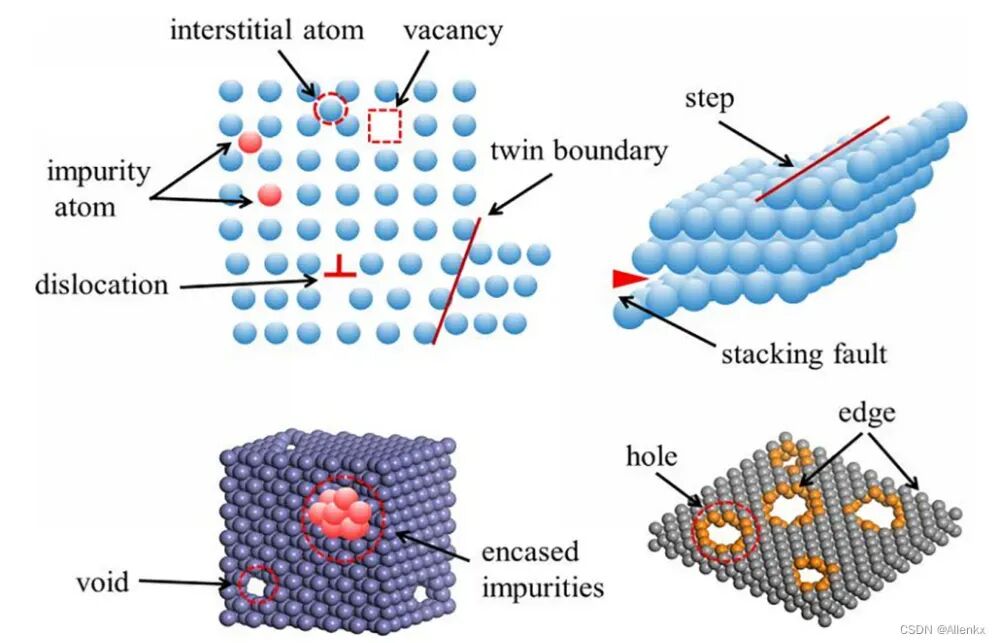
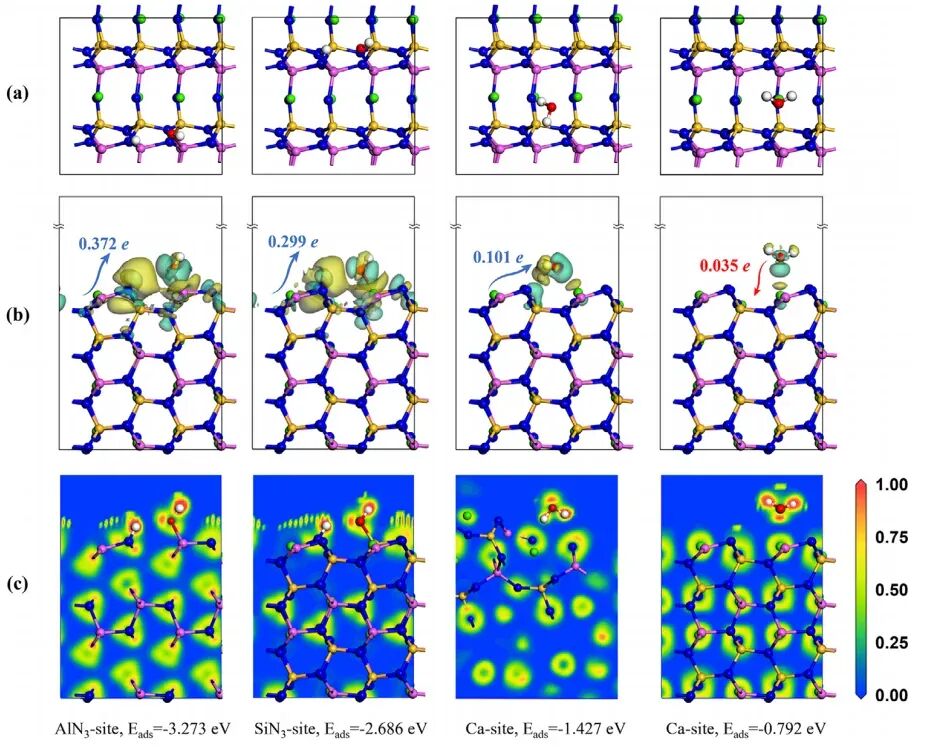
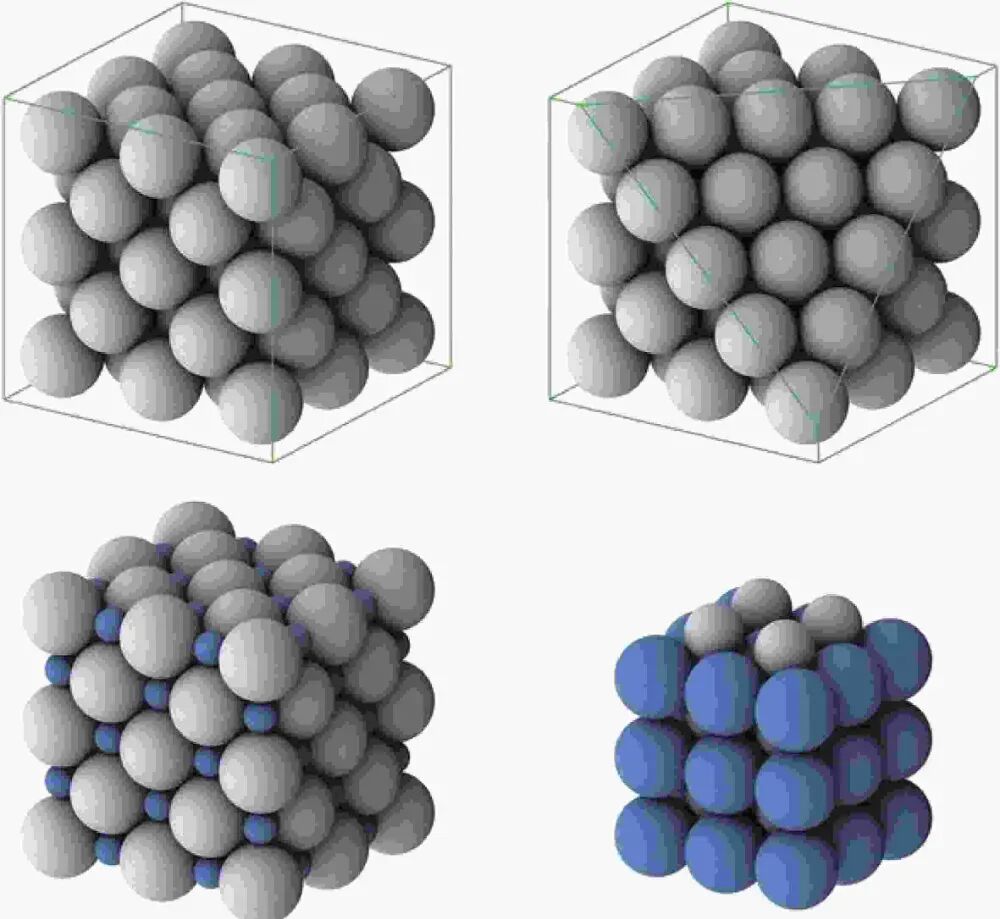
VASP(Vienna Ab initio Simulation Package)是一款广泛应用于材料科学和计算化学领域的第一性原理计算软件,广泛用于模拟固体、表面和界面的物理性质。
在使用VASP进行表面能计算时,用户常常会遇到一系列问题,这些问题可能涉及模型构建、收敛性、参数设置、计算精度等方面。华算科技朱老师将从常见问题及其解决方案的角度进行详细分析。
在进行表面能计算时,模型的构建和结构优化是关键步骤。常见的问题包括:
例如,表面模型的层数不够厚,导致表面能计算结果不稳定。例如,某些表面材料在计算时如果层数不够多,即使设置ISIF=2(仅弛豫原子位置),也可能导致表面结构“跑开”。
此外,模型构建时需注意真空层的设置,以避免不同表面单元之间的相互作用。
在进行表面弛豫时,需合理设置弛豫原子层。例如,固定中间层原子,观察层间距变化,若变化较大,则需增加弛豫原子层数。
此外,结构优化过程中需确保收敛性,例如通过调整EDIFF和EDIFFG参数控制电子和离子收敛。
表面能的计算通常涉及体相能量(E_bulk)和表面相能量(E_slab)的计算。用户可能不清楚如何从输出文件中提取这两个能量值。
例如,E_bulk通常通过固定所有原子的结构优化得到,而E_slab则通过弛豫后的表面模型计算得到。
例如,截断能(ENCUT)、k点采样(KPOINTS)、收敛标准(EDIFF、EDIFFG)等参数设置不合理,可能导致计算结果不收敛或精度不足。
例如,截断能设置过低可能导致计算精度下降,而k点采样不足可能导致收敛性差。
在计算过程中,可能遇到能量漂移、振荡或负值等问题。例如,负电荷密度的出现可能与ISMEAR设置不当有关,可通过调整LPARD、FFT网格等参数进行优化。
在大规模计算中,VASP的性能可能受到硬件限制,例如内存溢出或并行计算效率低等问题。例如,在SGI Origin等特定硬件上,可能需要调整临时文件设置(如IWAVPR)以提高性能。
交换关联函数(如LDA、GGA)的选择对计算结果有显著影响。例如,Perdew-Wang 91(GGA=91)和Perdew-Burke-Ernzerhof(GGA=PE)等泛函的选择需根据材料特性进行调整。
对于过渡金属表面,自旋极化设置(ISP)和初始磁矩设置对计算结果有重要影响。
计算结果的准确性需通过实验数据或理论模型进行验证。例如,通过比较不同方法(如Hartree-Fock)的计算结果,评估VASP计算的可靠性。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!