

说明:本文华算科技简要介绍了原位同步辐射X射线吸收光谱(XAS)在电催化研究中的应用,重点解析XANES与EXAFS在识别催化剂电子结构与局部配位环境中的作用,并展示其在揭示电催化反应动态过程中的独特优势。
XAS光谱可以被划分为两个区域,即XANES和EXAFS(图1)。XANES光谱表示了从核心能级到未占据态的电子跃迁的显著特征,反映了边前轨道(例如,氧化态)的电子结构,涉及电催化中的反应物和中间体杂化态。
此外,由于多重散射对XANES中产生的光谱特征贡献很大,XANES光谱也能提供有关被探测原子的配位几何结构的信息。
EXAFS光谱源于邻近原子的反散射光电子引起的干涉,是吸收信号在扩展边的振荡部分。EXAFS因此能够揭示吸收原子周围的局部配位环境(即,配位元素、配位数和原子间距离)。
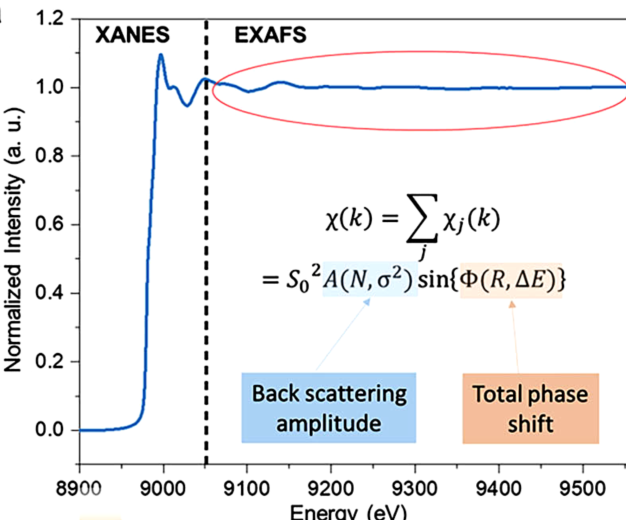

图1:典型的XAS光谱,插图是EXAFS光谱的定量参数化
(DOI:10.1038/s41467-023-42370-8)
对于XANES区域,一般来说,吸收边的能量可以提供氧化态的“指纹分析”。一般,XANES光谱的第一导数的峰位被用来直接识别Eedge(图2左)。
注意,由于边前区域可能存在电子激发,这种方法可能会导致Eedge位置的识别模糊。为了避免这种情况,使用归一化XANES光谱的半高位置(0.5)作为Eedge(图2右)。
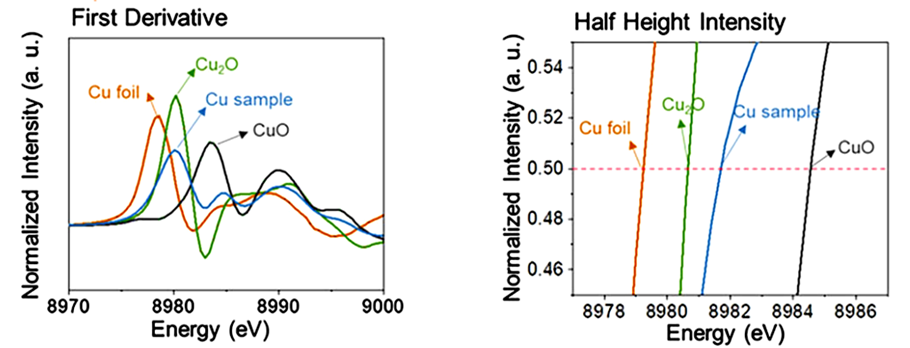
图2:一阶导数(左图)和半高宽强度(右图)
(DOI:10.1038/s41467-023-42370-8)
另一种方法是以积分的方式定义Eedge的平均值(图3),假设所有数据点在一个精选区域内,该区域排除了光谱噪声和边前特征(μ1和μ2之间的间隔)。
研究发现,积分方法相较于半高和导数方法更为精确,因为它能够合理处理复杂的边形状,考虑了多个数据点的高精度,并且对平滑处理不敏感。
注意:积分法也有局限性,它依赖于两个参数(μ1和μ2)的选择,并且对归一化的准确性有较高要求。
在评估氧化态时,必须特别注意XANES区域中显著的多重散射效应,因为吸收原子周围的不同配位结构和对称性会在XANES光谱的形状上产生明显差异,这可能导致在确定Eedge位置时出现歧义。
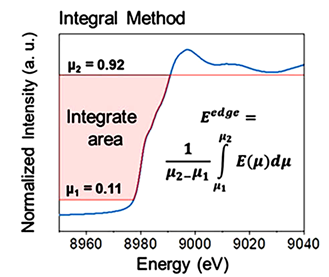
图3:积分法定义Eedge的平均值
一些研究表明,某些金属的氧化态即使相同,吸收边的位置和形状也会受到吸收位点几何细节的显著影响。因此,要准确从XANES光谱中识别氧化态,需要参考具有相似配位结构且无显著对称性变化的样品。
对于非均匀系统中可能存在的多种组分,传统方法难以精确确定氧化态。这时,线性组合分析(LCA)可以派上用场,它通过将实验XANES光谱与已知组分光谱的线性组合进行拟合,定量分析各组分的比例。
LCA利用已知结构的参考材料XANES光谱进行数据拟合,有效考虑了多重散射效应,从而提高了氧化态分析的准确性。
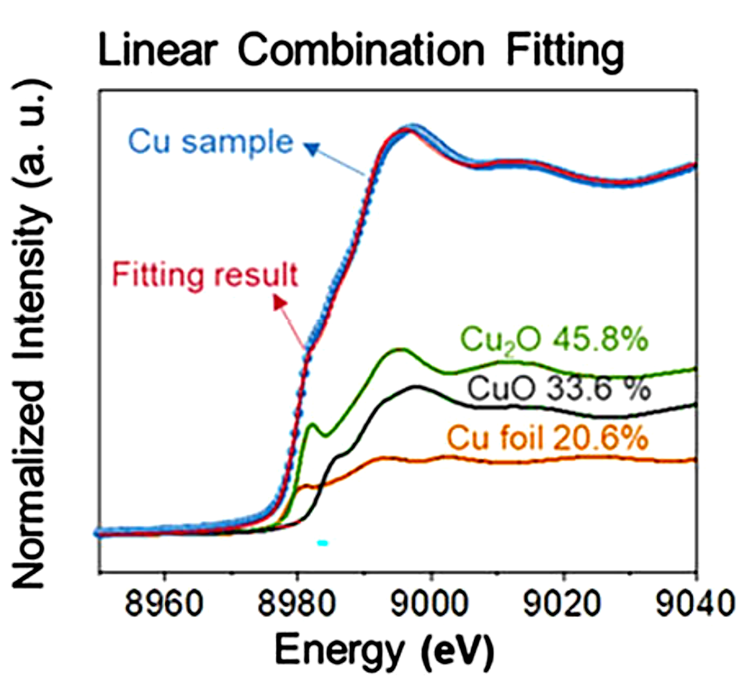
图4:线性组合拟合
EXAFS光谱通过将能量(E)转换为光电子波数(k)来获得,并通过傅里叶变换(FT)从k空间转换到R空间。
通过将得到的R空间光谱与参考材料的光谱进行定性比较,可以初步解析EXAFS数据。除了传统的FT-EXAFS方法外,小波变换(WT)提供了一种新的分析途径,它不仅揭示了光谱中存在的原子间距离,还实现了R和k空间的二维可视化,尤其适用于区分具有相似距离的多条散射路径。
例如,在基于铜(Cu)的混合物样品中,WT-EXAFS光谱清晰地区分了Cu-Cu散射路径,表明它们可能源自铜金属和氧化物。
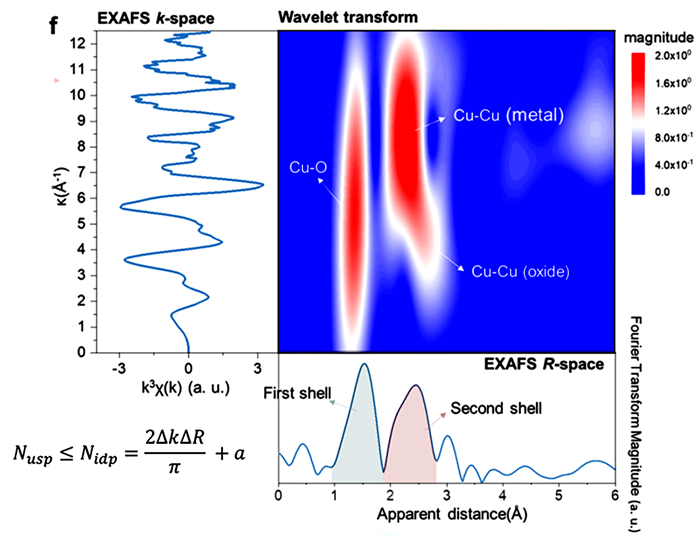
图5:基于铜的混合样品的Cu K边EXAFS光谱及其FT和WT
在电催化过程中,电催化剂表面的几个原子层对催化效果起着决定性作用。XAS光谱可以有效地识别关键中间体吸附后固液界面的原子构型。
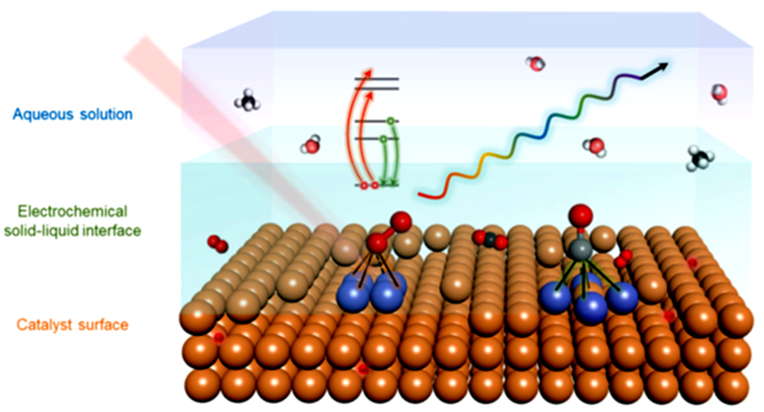
图6:分子层面上的电化学固液界面的示意图
Tran – Phu 等人通过 XANES 分析发现,在 1 M KOH 中,Co₃O₄ 会自发转变为 CoOOH,且 Co 氧化态略有升高,这意味着 Co 被还原为 Co (OH)₂。
EXAFS 结果显示,与新鲜催化剂相比,八面体(Oh)和四面体(Td)位点均存在 Co – Co 键合,不过 Td 位点减少。在 -0.52 V 时,Co (Oh) 峰增强、Td 峰减弱,表明无序度增加,这是 HER 性能提升的原因。
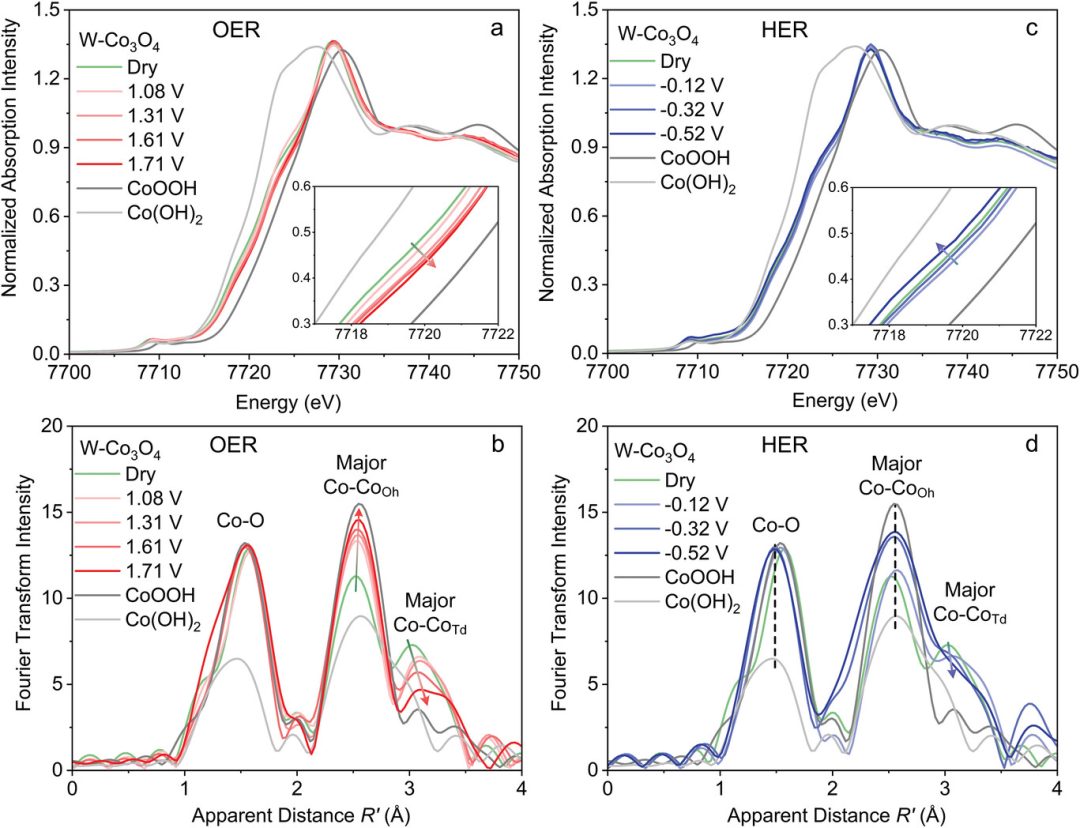
图7:在 1 mol/L KOH 电解液中施加 a、c)阳极和 b、d)阴极电位时 W-Co₃O₄的原位 XAS 光谱,以及 CoOOH、CoO 和 Co (OH)₂参比物的 XAS 光谱。
(DOI: 10.1002/smll.202208074)
除阳离子掺杂外,阴离子掺杂也会影响催化剂动态过程。Fan 等人以碳布负载的 S 掺杂 Co₃O₄(S – Co₃O₄/CC)为模型研究其 HER 演化。
原位 Co K 边 XANES 谱表明,0 mV 时 Co³⁺初步还原为 Co²⁺,-50 mV 时无进一步结构变化;而在 -200 mV 时,Co²⁺特征峰强度持续显著下降,进而形成 Co⁰。
原位表征共同显示,改善 HER 活性的活性位点是金属 Co 表面形成的 CoSₓ 纳米颗粒,凸显 “金属表面附着颗粒” 体系是该催化剂的关键特征。
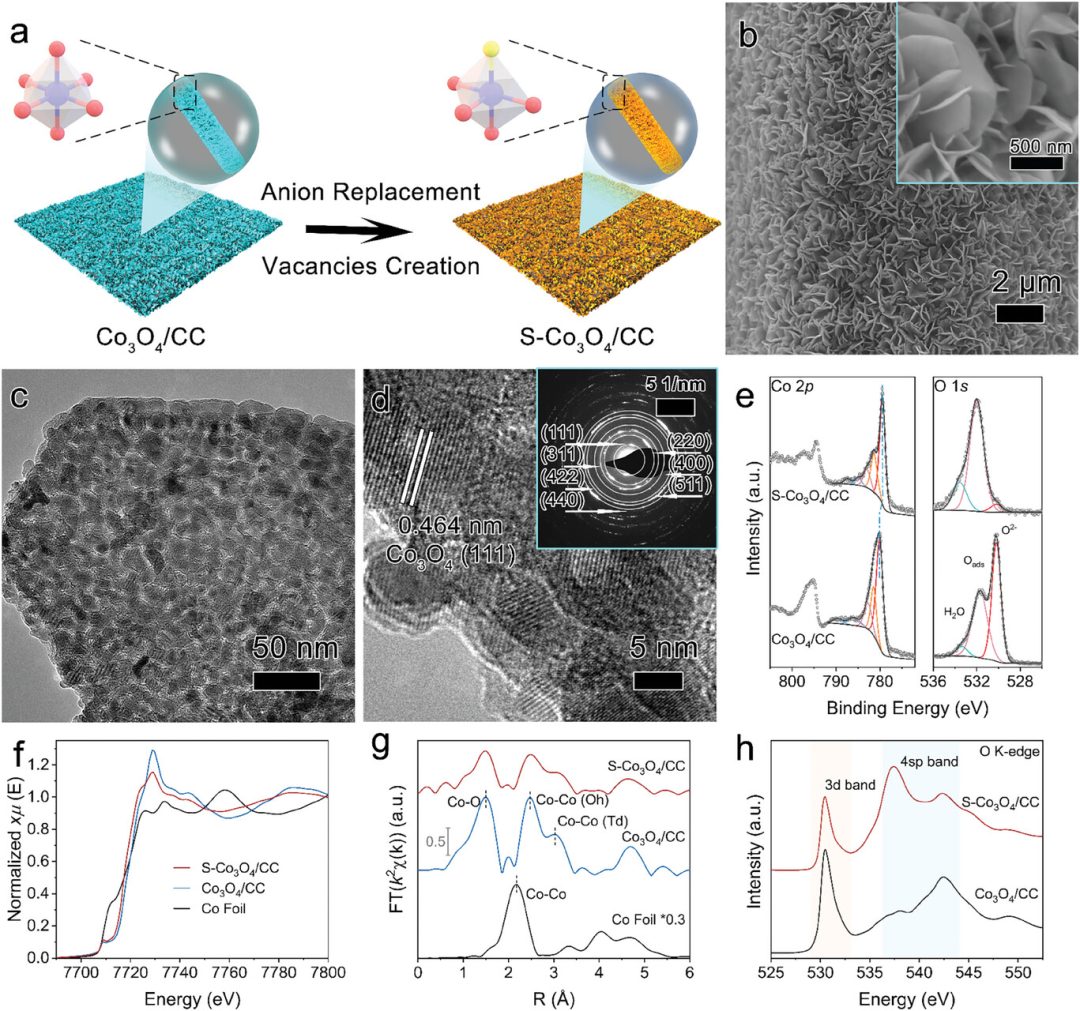
图8:(a)通过硫化氢气相光还原(H₂S – VPH)法制备 S – Co₃O₄/CC 过程中,Co 原子配位几何结构演变的示意图。(b)S – Co₃O₄/CC 的扫描电子显微镜(SEM)图像,(c)透射电子显微镜(TEM)图像,(d)高分辨率透射电子显微镜(HR – TEM)图像。(e)S – Co₃O₄/CC 和 Co₃O₄/CC 的 Co 2p X 射线光电子能谱(XPS)与 O 1s XPS 光谱。(f)S – Co₃O₄/CC、Co₃O₄/CC 和 Co 箔的 Co K 边 X 射线近边吸收精细结构(XANES)光谱,(g)傅里叶变换扩展 X 射线吸收精细结构(FT – EXAFS)光谱。(h)S – Co₃O₄/CC 和 Co₃O₄/CC 的 O K 边 XANES 光谱。
(DOI: 10.1002/aenm.202400052)
Li 等人利用原位 XAS 研究氧空位对 Rh 基纳米颗粒上 SLNP – RhO₂ 团簇耐久性的影响:测试 1 小时后,Rh L 边谱向低能移动 9 eV,与 Rh 纳米颗粒还原后进入下一阶段一致;初始 1 小时后,Rh – O 键合比例小幅下降后保持稳定,说明催化剂先部分还原,随后维持该状态。
相比之下,无空位的商业 RhO₂ 在 HER 中未向金属态转变,体现氧空位能显著增强催化剂稳定性。
在另一项研究中,Li 等人构建了含分离氧空位的 V₀ – Ru/HfO₂ – OP 催化剂,原位 Ru K 边 XANES 谱显示 Ru 纳米颗粒在 HER 中发生可逆氧化还原反应,Ru 价态随施加电位变化;
EXAFS 谱表明 HER 过程中 Ru – O – Hf 和 Ru – Ru 键长与强度呈可逆变化,说明氧空位诱导的灵活结构使该催化剂能承受结构畸变且不影响稳定性。
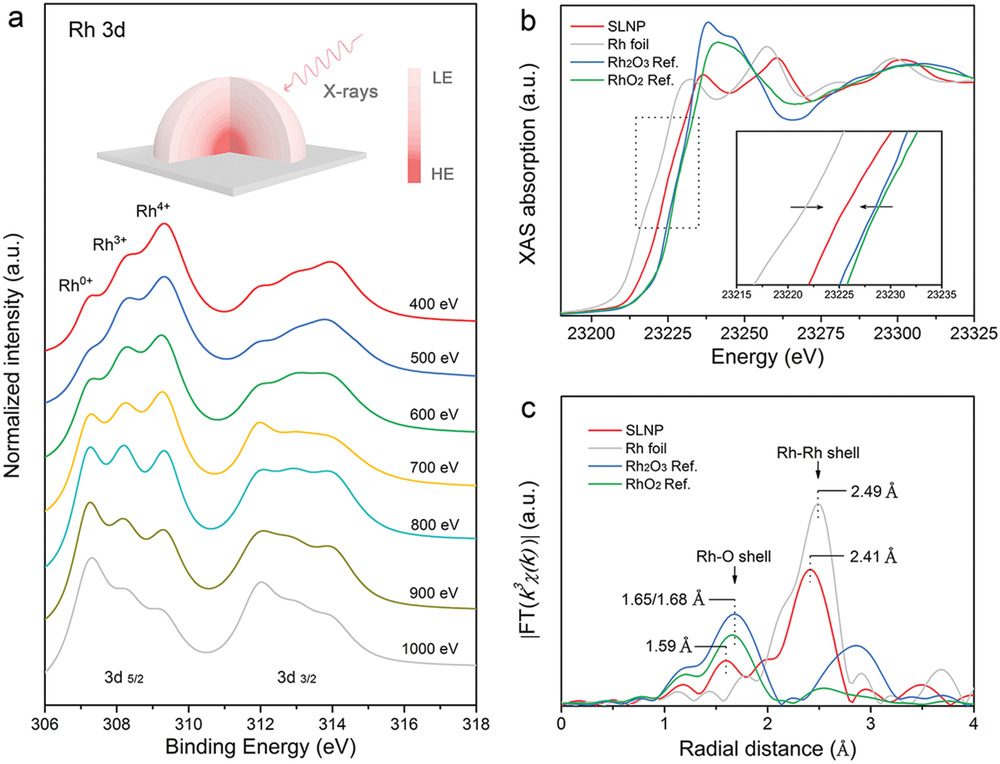
图9:SLNP 催化剂表面与体相的同步辐射表征。(a)分别在入射 X 射线能量为 400、500、600、700、800、900 和 1000 eV 时记录的 SLNP 同步辐射光电子能谱(SRPES)。插图为通过改变 X 射线辐射能量进行 SRPES 记录的示意图,LE 和 HE 分别代表低能和高能。(b)在 SLNP 的 Rh L 边以及标准 Rh 箔、Rh₂O₃和 RhO₂参比物处记录的体相归一化 X 射线近边吸收精细结构(XANES)光谱。(c)SLNP、Rh 箔、参比物 Rh₂O₃和参比物 RhO₂在 Rh L 边处的扩展 X 射线吸收精细结构(EXAFS)光谱的体相归一化傅里叶变换(FT)k³ 加权 χ(k) 函数。
(DOI: 10.1002/adma.201908521)
Pattengale 等开发了用于 HER 的 Ni@1T – MoS₂电催化剂,并通过 Mo 和 Ni K 边原位 XAS 研究其活性位点。他们发现,Mo 位点保持稳定,而 Ni 单原子位点变化显著。
在酸性条件下,Ni 中心的氧化态无结构改变(图 8b);在碱性条件下,配位数降低促使 NiSₓ物种形成以及可逆金属 Ni 活性位点产生。
研究证实,孤立的 Ni(Ⅱ)原子在酸性条件下有活性,且能形成催化活性 Ni⁰物种,结果表明 Ni@1T – MoS₂基面的 Ni 位点在酸性条件下对 HER 有活性,在碱性介质中会发生功能演化。
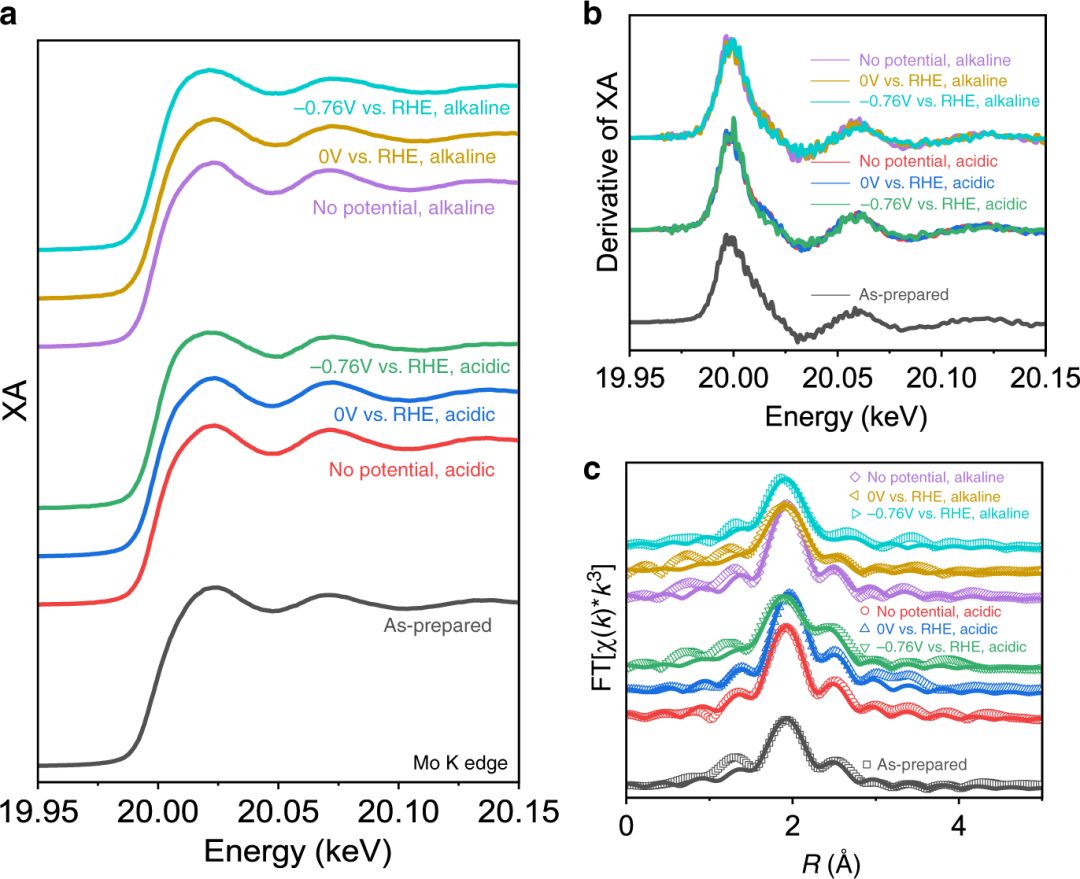
图10:(a)Ni@1T-MoS₂的X射线近边吸收精细结构(XANES)光谱,(b)一阶导数光谱,(c)傅里叶变换R空间光谱(空心点)及拟合曲线(实线)。
(DOI: 10.1038/s41467-020-17904-z)
XAS中的K边跃迁(1s → np)主要用于揭示原子间的距离和配位数等局部结构信息,但由于过渡金属的前轨道主要由d轨道构成,对催化反应的敏感性较低。
相比之下,L边跃迁(2p → (n-1)d)能直接揭示参与化学反应的未占据d轨道电子结构,是观察活性金属中心与吸附物化学键合的关键。
第一排过渡金属催化剂通常涉及2p到3d的L边跃迁,这在软X射线区域()发生。由于软X射线在环境或液体中的传播距离短,相关实验多在超高真空(UHV)条件下进行。
软X射线光谱学是研究电催化剂中轻元素(如C、N、O)的重要手段,这些元素的K边电离能位于200到1000 eV之间,常涉及反应物、中间体和产物。
原位探测这些元素能提供催化剂动态结构的关键信息,以及电解质与催化剂–吸附物相互作用的动态结构,对于理解催化过程至关重要。
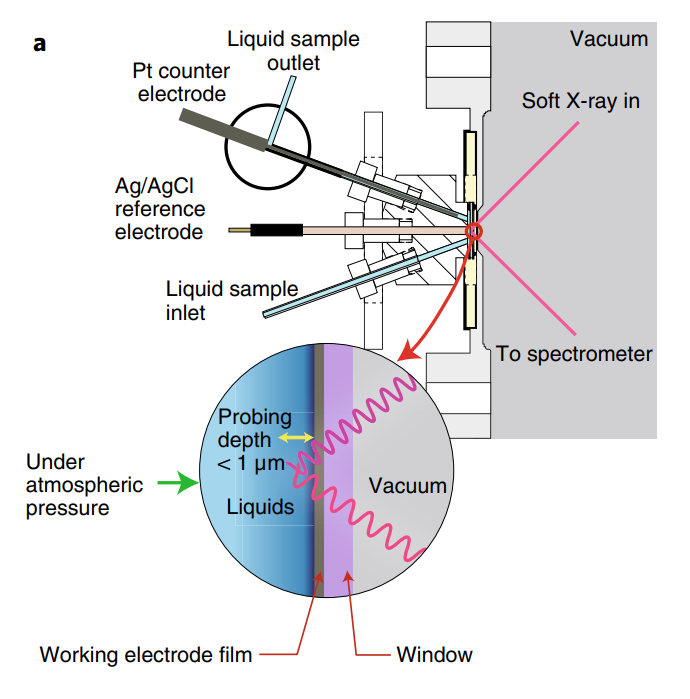
图11:软X射线工作原理示意图
(DOI:10.1038/s41467-023-42370-8)
D. Drevon通过原位XAS技术,研究了Ni-Fe(OxHy)电催化剂中氧原子的局部键合状态和对称性特征。O K边光谱显示了两个区域:525-534 eV区域指示O(1s)到O(2p)与金属(M)3d轨道的杂化,534-540 eV区域与水分子相关。
529、529.9、531.2和532.5 eV处的峰分别对应O到Ni(3d)t2g、Fe(3d)t2g、O2的π*和Fe(3d)eg的跃迁。529 eV处的峰在1.48 V vs RHE时增强,与Ni2+/Ni3+氧化还原一致,表明Ni(3d)与O(2p)杂化增强,促进电子重新分布,提高OER活性。
529.9和532.5 eV处的峰变化较小,显示Fe位点的调制有限。碳K边XAS技术区分了CO2、CO和还原碳形式,揭示了CO2RR中的关键中间体和结合模式,如CO2在CeO2 (110)上的不同吸附形态,以及CO在Au(111)位点的强结合,证实了XAS技术在识别轻元素化学键合方面的能力。
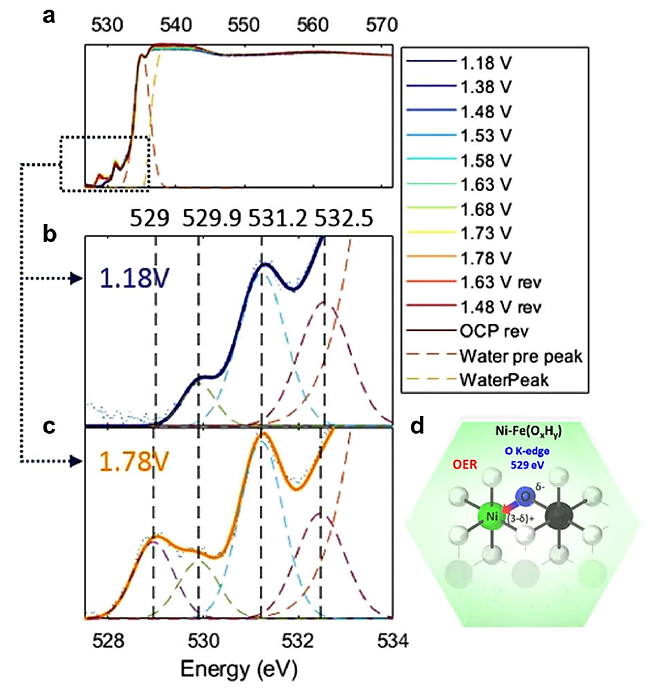
图12:在碱性OER期间,通过电沉积制备的Ni-Fe电催化剂的原位软X射线XAS在O K边的光谱
(DOI: 10.1038/s41598-018-37307-x)
Marcel Risch使用Mn L3,2边XAS光谱证明了在OER和ORR相关电位下沉积的锰氧化物薄膜的动态氧化态。
在高于1.50 V相对于RHE的电位时,光谱与参考谱匹配得很好,在低于0.80 V相对于RHE的电位时,光谱与含有四面体位点Mn2+的Mn3O4参考谱一致。
这些发现表明,锰氧化物在ORR期间包含四面体Mn2+位点,而在OER期间特征是混合的Mn3+/4+价态。基于Mn在循环电位期间的氧化还原行为,可以进一步推断出锰氧化物的氧化动力学比还原动力学更缓慢。
此外,与硬X射线相比,软X射线XAS被认为是表面敏感的,电沉积的Cu氧化物在CO2RR期间的动态表面氧化态也可以通过原位Cu L边XAS测量来探测。
通过对原位Cu L边XAS光谱的线性组合分析表明,在-1.2 V相对于RHE的初始2分钟电催化中,表面Cu物种由84%的Cu+组成,然后在接下来的10分钟内Cu+的量减少到77%。
在1小时CO2RR电催化后,仍有23%的Cu+物种存在,这被认为是高效CO2到乙烯转化的原因。
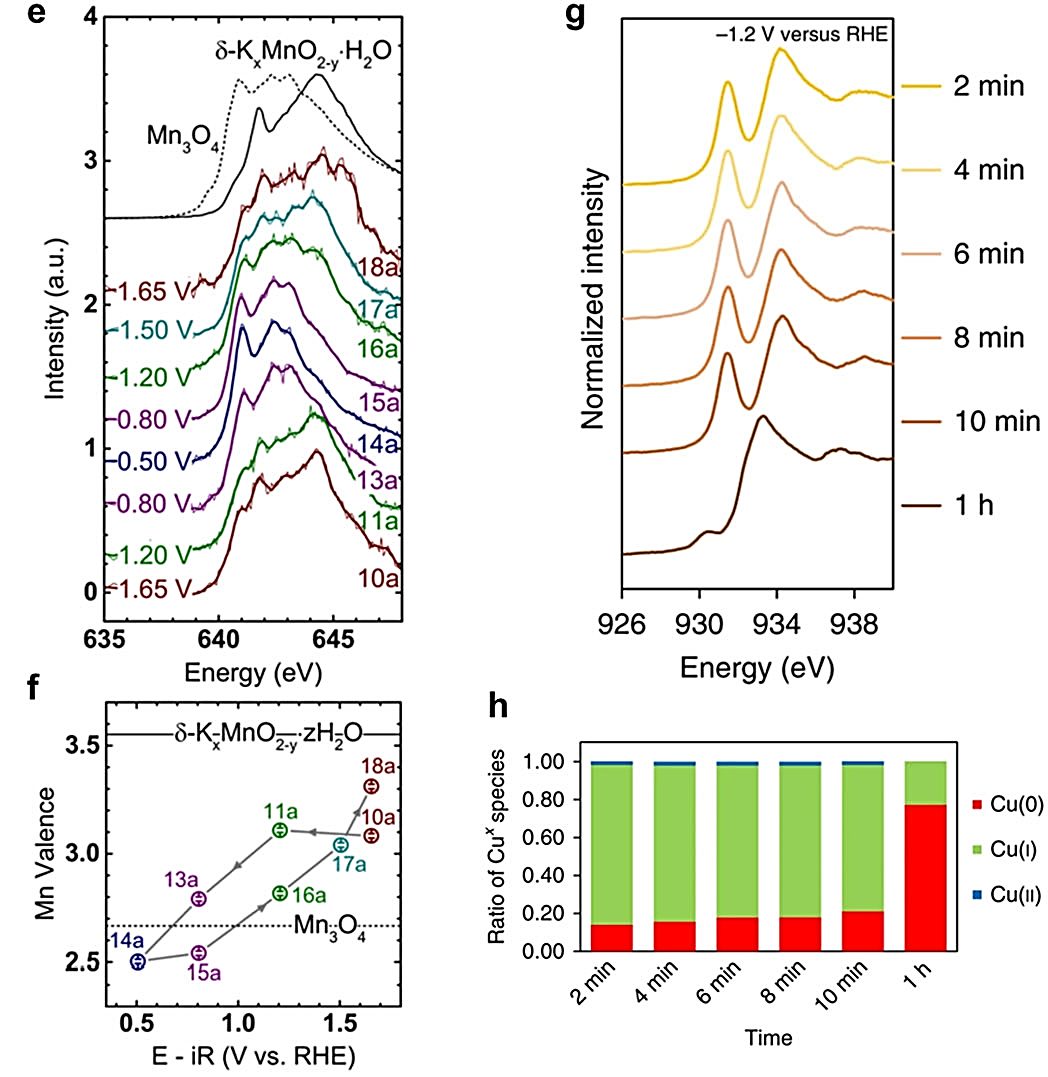
图13:荧光模式记录的电沉积锰氧化物在Mn L3,2边的原位软X射线XAS光谱
(DOI: 10.1021/acs.jpcc.7b05592)
原位XAS技术为深入理解电催化剂的活性位点、结构演化与反应机制提供了强有力的工具,尤其在捕捉催化过程中的瞬态结构变化方面展现出不可替代的价值。