

说明:本文将围绕简单小分子DFT 计算展开,先界定其基于 HK 定理、针对原子数50 分子的定义与优势;再讲 “体系定义-参数选择-计算执行-结果验证”完整流程,含泛函基组选择等;还述及四大应用场景、五大注意事项,最后以顶刊案例佐证,全面呈现其理论与应用价值。
什么是简单小分子的DFT 计算
密度泛函理论(DFT)是计算量子化学的核心方法,通过求解电子密度分布而非波函数,高效预测分子结构、能量及反应性质。简单小分子指原子数<50的有机/无机分子(如H2O、苯、氨基酸),其DFT计算可在普通工作站完成,适用于:几何优化(键长/键角)、电子性质(轨道能级、偶极矩)、弱相互作用能(氢键、范德华力)等。
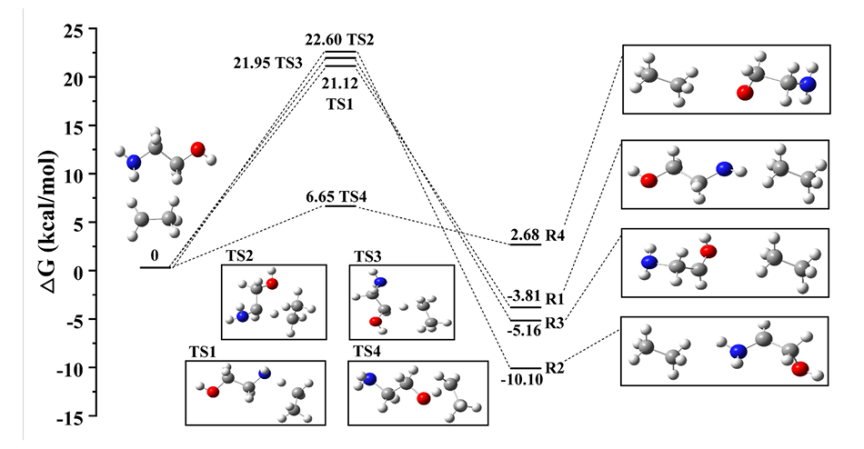

DOI:10.1016/j.ces.2024.120458
相比于基于波函数的量子化学方法(如Hartree-Fock、MP2、CCSD(T)等),DFT具有计算效率高、精度适中的特点,使其能够处理包含数百甚至上千个原子的体系,在计算化学、材料科学和表面催化等领域得到了广泛应用。
DFT计算的核心流程
计算前需要明确体系与基础参数,在进行简单小分子的DFT计算时,通常包含几个关键步骤:基组选择、泛函选择、几何优化、性质计算和结果分析。每个步骤都需要根据研究体系和目标性质做出适当的选择,这对计算结果的准确性和可靠性至关重要。
明确体系与基础参数
首先需定义小分子的核心参数,避免因初始设置偏差导致结果无效。核心参数包括电荷与自旋多重度——简单小分子多为电中性(电荷= 0),自旋多重度需根据未成对电子数确定:如H2(无未成对电子,多重度= 1,单重态)、O2(2个未成对电子,多重度= 3,三重态)、NO(1个未成对电子,多重度= 2,二重态)。
其次是分子初始结构构建——需基于实验数据或合理猜想搭建初始构型,避免初始结构与极小点偏差过大导致计算不收敛。
例如构建 H2O分子时,初始键长可设为 0.96 Å(实验值0.958 Å),键角设为 104.5°(实验值104.7°);
构建 CO2时,初始C=O 键长设为 1.16 Å(实验值 1.163 Å),键角设为180°(线性分子)。可通过GaussView、VESTA 等软件可视化构建,部分软件支持直接导入实验晶体结构(如从 CCDC 数据库获取小分子晶体信息)。
基组和泛函选择
基组(Basis set)是用于描述分子轨道波函数的数学函数集,其选择对计算精度和效率有直接影响。
常用的基组包括分裂价壳基组(如6-31G、6-311G)、极化基组(添加角动量函数,如6-31G(d)、6-311++G(d,p))和扩散函数基组(用于描述远离原子核的电子,如aug-cc-pVDZ)。
对于小分子计算,通常建议使用至少三重zeta质量的基组并包含极化函数,如6-311++G(2d,p)基组,它在计算成本和精度间提供了良好平衡。
交换-关联泛函的选择是DFT计算中最关键的决策之一。常见的泛函类型包括:局域密度近似(LDA)、广义梯度近似(GGA,如PBE、BLYP)、meta-GGA(如TPSS、SCAN)和杂化泛函(如B3LYP、PBE0、HSE06)。
杂化泛函由于包含一定比例的精确Hartree-Fock交换能,通常能提供更精确的结果,特别是对于分子能隙、反应能垒等性质的预测。
计算执行:参数设置与任务类型
简单小分子的DFT计算任务主要分为三类,需根据研究目标选择对应的计算关键词(以 Gaussian软件为例子)。
(1)几何优化(Opt)
目标是找到分子的能量极小点(稳定构型),关键词为“# B3LYP/6-311G (d,p) Opt”。计算过程中,软件会迭代调整键长、键角,直至能量梯度小于设定阈值(默认0.0015 hartree/Å)。
需注意:优化完成后需检查是否存在虚频(振动频率为负值)——若有虚频,说明结构为过渡态或不稳定构型,需重新调整初始结构。
(2)能量计算(SP)
目标是获取优化后分子的单点能(如键解离能、结合能),关键词为“# M06-2X/def2-TZVP SP”。需基于优化后的稳定结构进行,避免直接对初始结构算单点能(误差可能超 100 kJ/mol)。
例如计算H2O的O-H键解离能,需先优化H2O、OH・、H・的结构,再通过“E (OH・) + E (H・) – E (H2O)”计算解离能,结果约为498 kJ/mol,与实验值502 kJ/mol 误差 。
(3)电子性质与振动频率(Freq、Pop)
振动频率(Freq):关键词为“# B3LYP/6-311G (d,p) Opt Freq”,可预测分子的红外(IR)、拉曼光谱,用于与实验谱图对比验证。
例如CO2的计算红外光谱中,对称伸缩振动峰位于1330cm-1,反对称伸缩峰位于2349cm-1,与实验谱图完全吻合;
电子性质(Pop=NBO):通过自然键轨道(NBO)分析获取电荷分布、HOMO-LUMO能级。例如 H2O的NBO电荷分布中,O原子带 -0.82 e,H 原子带+0.41 e,与实验测得的偶极矩(1.85 D)一致;
HOMO能级为 -12.6 eV,LUMO 能级为-4.8 eV,可用于判断分子的反应活性(HOMO越低,分子越稳定)。
DFT计算在简单小分子中的应用领域
DFT计算在简单小分子的研究中有着广泛的应用,几乎涵盖了化学、材料和生物学的各个领域。通过这些计算,研究人员可以在分子水平上理解化学反应的机理、预测材料的光电性质以及设计具有特定功能的新分子。
在催化领域,DFT被广泛用于研究小分子在催化剂表面的吸附和活化机制。
例如,研究人员通过DFT计算研究了NH3、NOx和O2小分子在Fe3O4(111)表面的吸附行为和活化规律,为理解氨选择性催化还原(NH3-SCR)反应机理提供了原子水平的见解。
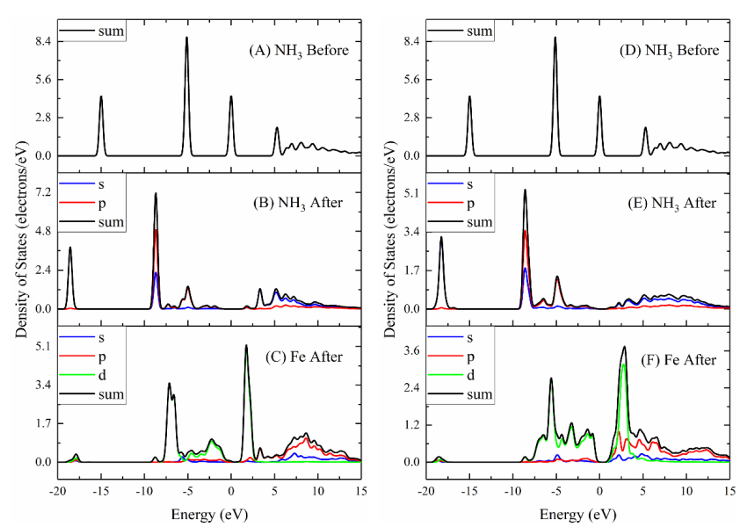
图1 NH3吸附在Fetet和Feoct-tet端表面前后最稳定构型的相关原子或分子的DOS和PDOS
DOI:10.1016/j.apsusc.2022.154645
计算表明,NH3主要通过N原子与表面的Fe位点(Fetet或Feoct)形成化学键,吸附能在-0.27 eV到-1.48 eV之间,而NO则可以通过N端或O端吸附,形成不同的吸附构型
在光谱预测方面,DFT计算可以准确模拟分子的红外(IR)、拉曼(Raman)、紫外-可见(UV-Vis)光谱以及荧光光谱等,辅助实验光谱的指认和解析。
通过计算分子的振动频率和强度,可以获得与实验光谱对比的理论光谱,从而识别分子的特征峰和结构特征。
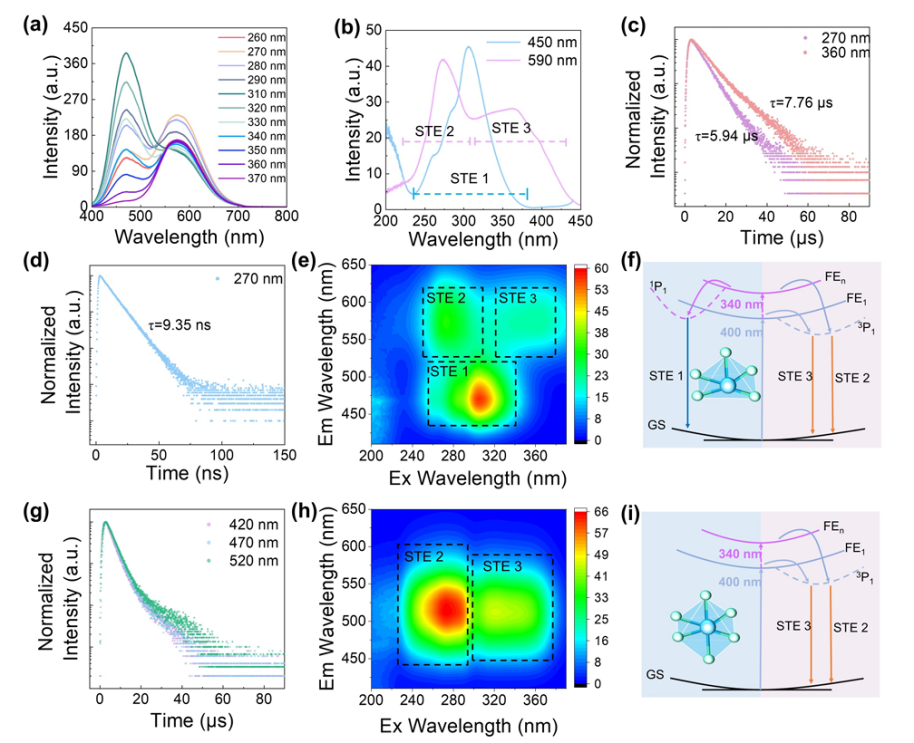
图2 DFT计算光谱模拟
DOI: 10.1002/anie.202412253
在材料科学领域,DFT计算为有机太阳能电池(OSCs)和光电材料的设计提供了理论基础。
研究人员通过DFT计算设计了一系列基于苯并二噻吩(BDT)、噻吩并[3,2-b]噻吩(TT)和Ullazine核的小分子给体材料。
通过计算分子的前线分子轨道(HOMO/LUMO)、激发能、重组能和吸收光谱等性质,可以预测材料的光电转换效率,指导高性能材料的合成。
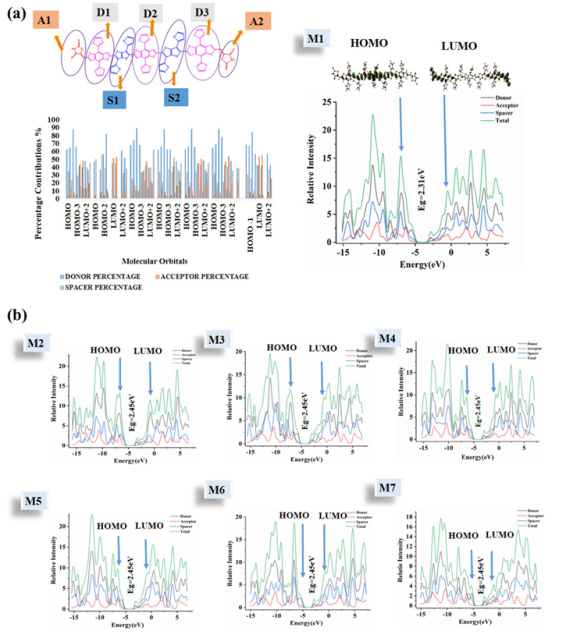
图3 DFT计算分子的前线分子轨道、激发能光谱模拟
DOI:10.1007/s10895-025-04446-0
例如,对Ullazine衍生物(UA1-UA6)的DFT计算表明,含氮/硫/羰基/氰基的UA6分子展现出最宽的吸收范围,UA1具有最优电子迁移率(2.19×10-3cm2V-1s-1),而UA4则凭借最高HOMO能级(-4.61 eV)和最低电离能(IP=5.71 eV),成为最佳空穴传输材料。
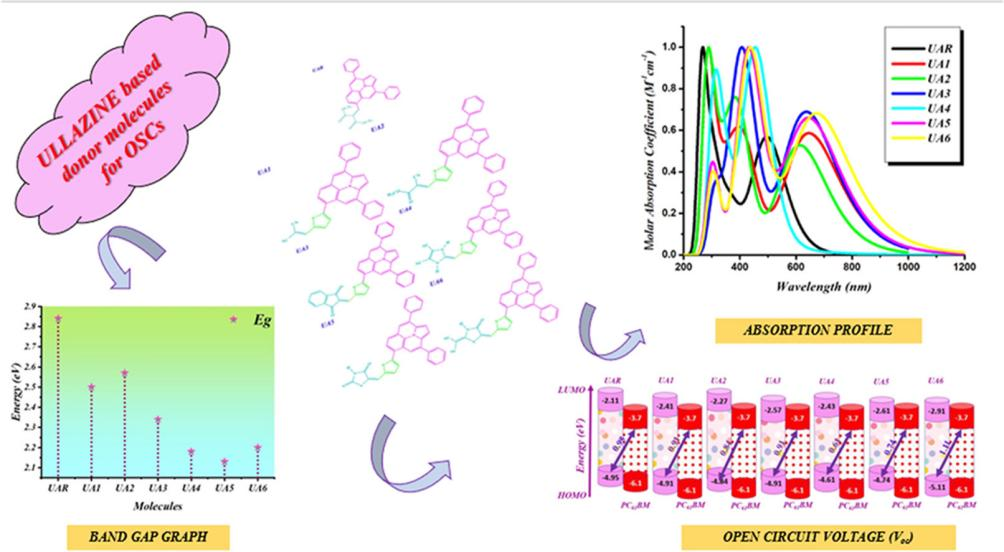
图4 Ullazine衍生物的前线分子轨道
DOI:10.1016/j.jmgm.2025.109112
在生物分子研究中,DFT计算可以用于研究小分子与生物大分子的相互作用,如药物分子与靶标蛋白的结合机制、抑制剂的设计与优化等。通过计算结合自由能、相互作用模式等参数,DFT计算可以为药物设计提供分子水平的见解,减少实验试错成本。
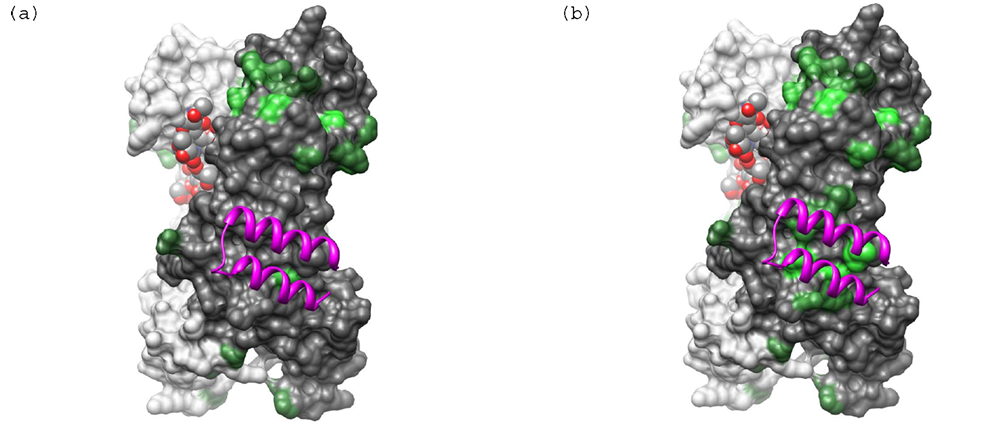
图5 小分子与生物大分子的相互作用能计算
DOI:10.1021/ja303612d
案例分析
DFT研究小分子表面吸附/活化规律
研究简介:该研究通过DFT计算系统地研究了NH3、NOx和O2小分子在Fe3O4(111)表面的吸附和活化行为,为理解氨选择性催化还原(NH3-SCR)反应机理提供了深入见解。
研究人员首先构建了Fe3O4(111)表面的两种终端结构:Fetet端面和Feoct-tet端面。Fetet端面由四面体Fe位点(Fetet)、孤立O位点(Oiso)、非孤立O位点(Onon-iso)和中空位点(Ohollow)组成;
而Feoct-tet端面则包括最外八面体Fe位点(Feoct)、次外Fetet和Oiso。通过比较不同吸附位点的吸附能和电子结构变化,他们发现了小分子在这些位点上的吸附规律。
对于NH3分子,研究表明它主要通过N原子与表面的Fe位点形成化学键5。在Fetet端面上,NH3垂直吸附在Fetet顶部,吸附能为-1.48 eV,N-Fe键长为2.109 Å,有0.04 e电子从NH3转移到表面5。
而在Feoct-tet端面上,NH3的吸附能较低(-0.70 eV和-0.27 eV),但通过水平吸附构型,NH₃的H原子与表面的Oiso之间形成氢键,有利于N-H键的活化和解离。
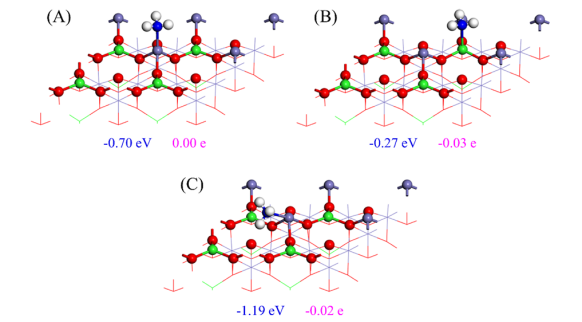
图6 平衡构型(A)在Feoct上(B)在Fetet上(C)在Oiso上,用于NH3在Feoct-tet 端表面上的吸附
DOI:10.1016/j.apsusc.2022.154645
进一步,研究人员通过过渡态搜索研究了NH3的氧化脱氢过程。发现由于Feoct-tet端面上NH3的水平吸附构型有利于形成氢键,其脱氢能垒(0.79 eV)远低于Fetet端面上的能垒(1.49 eV),表明Feoct-tet端面对NH3活化具有更高活性。
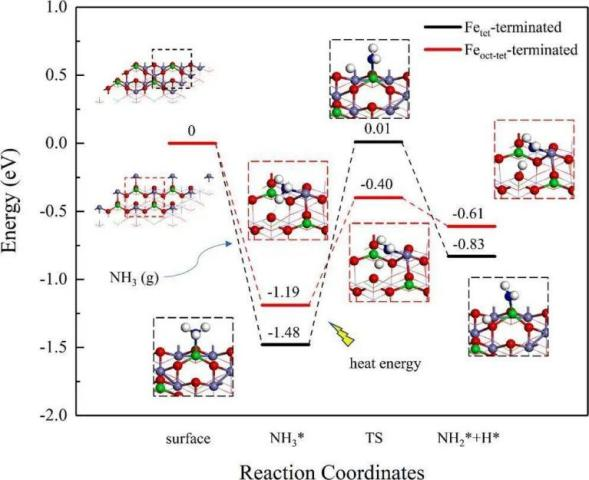
图7 NH3氧化脱氢反应在Fetet端表面(黑色)和Feoct-tet端表面(红色)上的势能图和优化结构
DOI:10.1016/j.apsusc.2022.154645
这一理论研究从原子级别解释了Fe3O4(111)表面的催化活性位点机制,为设计高效SCR催化剂提供了理论指导。
总结
DFT计算作为研究简单小分子电子结构和性质的重要工具,已经在化学、材料和生物等领域发挥了不可替代的作用。
通过不断改进的理论方法和计算软件,DFT计算能够以较高的精度和效率预测分子的结构、能量、光谱和反应性能,为实验研究提供深层次的机理理解和设计指导。