

XRD简介
XRD的基本原理
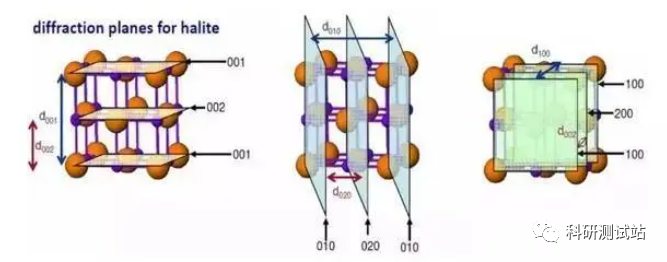

XRD样品要求与制样方法

样品要求:
(1)粉末样品要求:3克左右,如果太少也需5毫克,应干燥,粒度一般要求约10~80μm,应过200目筛子(约0.08mm),且避免颗粒不均匀。粒度粗大衍射强度底,峰形不好,分辨率低。要了解样品的物理化学性质,如是否易燃,易潮解,易腐蚀、有毒、易挥发。制样方法:用药勺取适量样品于玻璃样品架中间的槽里,另取一块载玻片,用载玻片轻轻将样品压紧,并将高出样品架表面的多余粉末刮去,如此重复几次使样品表面平整。将样品架边缘的样品刮掉,擦干净即可进行测试。注意事项:玻璃样品架易碎,使用要小心。制样时用力要均匀,不可力度过大,以免形成粉粒定向排列。样品一定要刮平,且与样品架表面高度一致,否则引起测量角度和对应d值偏差(样品平面高于凹槽平面会引起XRD峰的整体偏移)。
(2)其他样品样品要求:金属样品如块状、板状、圆拄状要求磨成一个平面,面积不小于10X10毫米,如果面积太小可以用几块粘贴一起。对于片状、圆拄状样品会存在严重的择优取向,衍射强度异常。因此要求测试时合理选择响应的方向平面。对于测量金属样品的微观应力(晶格畸变),测量残余奥氏体,要求样品不能简单粗磨,要求制备成金相样品,并进行普通抛光或电解抛光,消除表面应变层。

制样方法:
(1)对于不易研碎的样品,可先将其处理成与窗孔大小一致,磨平一面,再用橡皮泥或石蜡将其固定在窗孔内,固定时橡皮泥或石蜡等请注意略低于样品,不要与样品齐平。片状、纤维状或薄膜样品也可类似固定在样品架的窗孔内,应注意使样品表面与样品架平齐。
(2)对于不同基体的薄膜样品,要了解检验确定基片的取向,X射线测量的膜厚度约20个纳米。
(3)对于纤维样品的测试应该提出测试纤维的照射方向,是平行照射还是垂直照射,因为取向不同衍射强度也不相同。对于焊接材料,如断口、焊缝表面的衍射分析,要求断口相对平整。
XRD实验的注意事项

如果采用球磨等非常强力地方式将样品研磨的样品很细,可能会破坏晶型结构,且颗粒尺寸太小,会产生对X射线的吸收,衍射强度降低,晶粒尺寸小也会引起峰宽化,不利于得到结构清晰的XRD谱图。
XRD数据分析
Jade使用方法视频教程链接:https://pan.baidu.com/s/1BvbP8w-RhxRwmKSanUbRFA?pwd=1005 提取码:1005
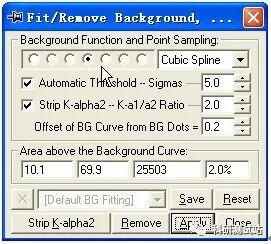
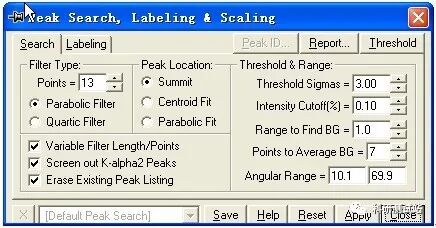
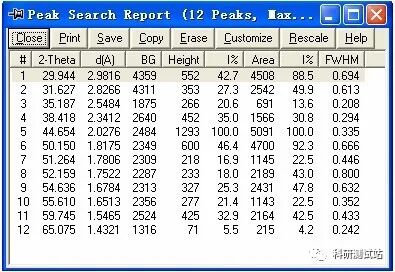
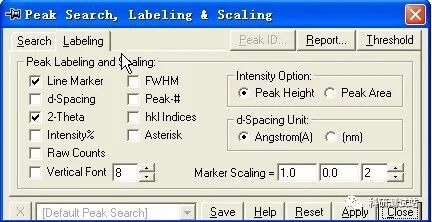
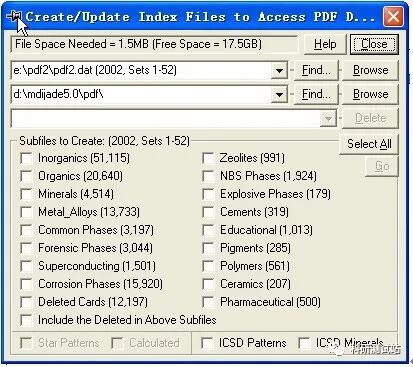
jade5.0 的基本操作步骤

XRD的应用
物相分析
物相分析是X射线衍射在金属中用得最多的方面,分定性分析和定量分析。前者把对材料测得的点阵平面间距及衍射强度与标准物相的衍射数据相比较,确定材料中存在的物相;后者则根据衍射花样的强度,确定材料中各相的含量。在研究性能和各相含量的关系和检查材料的成分配比及随后的处理规程是否合理等方面都得到广泛应用。
结晶度的测定
结晶度定义为结晶部分重量与总的试样重量之比的百分数。非晶态合金应用非常广泛,如软磁材料等,而结晶度直接影响材料的性能,因此结晶度的测定就显得尤为重要。测定结晶度的方法很多,但不论哪种方法都是根据结晶相的衍射图谱面积与非晶相图谱面积决定。
精密测定点阵参数
精密测定点阵参数 常用于相图的固态溶解度曲线的测定。溶解度的变化往往引起点阵常数的变化;当达到溶解限后,溶质的继续增加引起新相的析出,不再引起点阵常数的变化。这个转折点即为溶解限。另外点阵常数的精密测定可得到单位晶胞原子数,从而确定固溶体类型;还可以计算出密度、膨胀系数等有用的物理常数。
纳米材料粒径的表征
纳米材料的颗粒度与其性能密切相关。纳米材料由于颗粒细小,极易形成团粒,采用通常的粒度分析仪往往会给出错误的数据。采用X射线衍射线线宽法(谢乐法)可以测定纳米粒子的平均粒径。
晶体取向及织构的测定
晶体取向的测定又称为单晶定向,就是找出晶体样品中晶体学取向与样品外坐标系的位向关系。虽然可以用光学方法等物理方法确定单晶取向,但X衍射法不仅可以精确地单晶定向,同时还能得到晶体内部微观结构的信息。一般用劳埃法单晶定向,其根据是底片上劳埃斑点转换的极射赤面投影与样品外坐标轴的极射赤面投影之间的位置关系。透射劳埃法只适用于厚度小且吸收系数小的样品,背射劳埃法就无需特别制备样品,样品厚度大小等也不受限制,因而多用此方法。
XRD测试的常见问题
1. 为什么要求XRD测试粉末样品要稍微多一些?
因为粉末样品要铺满整个样品台,不然X射线可能会打在样品台上,影响数据质量。
2. 为什么XRD测试要求薄膜(块体)样品尺寸要合适?
因为放置薄膜(块体)样品的样品台尺寸是固定的,用橡皮泥来固定样品,样品太大放不进去,样品太小不好固定。
3. 为什么XRD数据的峰强度较低,甚至没有明显的衍射峰?
样品的衍射峰强度最主要跟样品本身的结晶度有关,其次跟样品量以及仪器的功率都有关系。
4.粉末样品量过少会有什么样的影响?
答:因为粉末样品要铺满整个样品台,样品量过少可能导致X射线打在样品台上,从而影响数据质量。
5.为什么部分样品的测试结果中衍射峰强度较低,甚至没有明显的衍射峰?
答:样品的衍射峰强度主要跟自身结晶度有关,结晶度越好,衍射峰越明显,其次才是样品量和仪器功率。
6.步长和扫描速度有何关联?扫描速度对样品的测量有何影响?
答:步长决定了扫描的精细程度,扫描速度是由步长和每步的时间决定的,每步的时间长短会影响衍射峰的强度和信噪比。每步的时间越长,步长越小,则谱越精细,信噪比越高,可用于定量计算,如XRD精修等。
7.XRD定量的精确度如何?
答:XRD为半定量分析,定量测试结果精确度有限,仅供参考。
8.为何有些样品的测试结果基线较高?能否改善?
答:有荧光散射现象的样品,如铁含量较高的样品,测试结果会出现基线较高的情况,这是无法改善的。
9.Cu靶的波长是多少?
答:Kα1=1.540538埃。
10.XRD掠入射角度如何选择?
掠射角度建议越小越好,一般都是0-1°最好,例如0.5°,不建议2°或3°,这种基本不叫掠射,基底的信号也会比较明显
11.XRD如何计算晶粒尺寸?
XRD计算晶粒尺寸需要知道峰的归属,如果可以对上标准谱,可以用卡片里的晶面指数代替,如果完全未知,需要先对谱图指标化,指标化的过程比较麻烦。
12.XRD小角掠入射测试深度是多少?
深度是一方面根据入射角来定的,另一方面还由样品的元素决定,入射深度可以进行计算,但是比较麻烦,元素的吸收系数都得考虑,通常情况下简单通过入射角可以粗略确定,如果要准确计算,需要查阅相关的文献资料。
13.薄膜状样品做XRD小角掠射只出来基底峰是什么原因?
一般膜层越薄,掠射角选取越小,一般是0.5-2度,理论上出基底峰的话也会有膜层的峰,但是如果膜层很薄只有几十纳米的话,即使掠射角做到0.5,出膜层衍射峰的几率也不大,即使出来也会被强大的基底峰掩盖住。
14.X射线单晶衍射仪和x射线多晶衍射仪有什么区别?
1)单晶衍射仪主要用于测定单个纯物质的晶体结构,对于已知结构,可以进行精修,对于未知结构,可以鉴定结构。要求所测的样品为块状单晶。一般在表征新化合物时,最好用单晶衍射仪,测量一个单晶体需要一到两天,解析一个单晶可能要花费更多的时间。另外,培养单晶也很不容易,单晶生长受很多条件的限制。2)多晶体衍射仪(XRD)也称为粉晶衍射,主要是用来测定样品的物相组成,它主要依据的PDF数据库,通过查找这个库中与样品衍射谱相同的物相来鉴定某个物相是否存在,因此,鉴定的必须是已知物相。也可以测量单晶,前提条件是把单晶破碎成粉晶,这时测量的相当于是纯物质。对于样品要求块状、粉末状都可以,样品容易制取,测量时间短,物相鉴定现对来说比较简单、快速。
15.XRD测试基线很飘的原因?
XRD测试数据基线飘是正常的,仪器光斑开的比较大,检测器窗口也大,这样的图谱信噪比和强度都会好很多,所以起始角度越低,前面的光子计数就越高。对数据分析没什么影响,不用刻意进行处理。
16.XRD小角测试应当注意的问题。
(1)如果看10度以下的峰,需要确认测试目的是看物相还是看孔结构;
(2)如果看物相,需要用广角模式测试,也就是测试5-90(或者3-90度都行),如果看孔结构,需要用小角模式,也就是0.5-10度;
也就是说,如果都是测试5-10度,用小角模式和广角模式,结果是不同的。
17.高温XRD随着温度升高,峰位为什么会出现左移现象?
随温度升高使衍射峰左移,这是因为一般材料都是正膨胀系数材料,即随温度升高而膨胀。根据布拉格公式2dsinθ= nλ,在波长不变的情况下,d值增大,必然使sinθ 变小,而在衍射范围内,也就是θ变小,即峰位左移。
本文源自微信公众号:科研测试站
原文标题:《【xrd知识大全】带你了解XRD基本原理、测试注意事项、数据分析及常见问题》
原文链接:https://mp.weixin.qq.com/s/dVmG1i3diYwMP3yUUez0oQ
本转载仅出于分享优质测试干货,旨在传递更多观点,并不代表赞同其全部观点或证实其内容的真实性。文章中所包含的图片、音频、视频等素材的版权均归原作者所有。如有侵权请告知删除。