VASP(Vienna Ab Initio Simulation Package)是一种广泛应用于材料科学和化学领域的第一性原理计算软件,尤其在电子结构计算、催化反应、材料性能预测等方面具有重要地位。
VASP的计算能力强大,但其使用和优化需要一定的技巧和经验。华算科技朱老师将围绕VASP计算HER(析氢反应)台阶图的高级技巧进行详细探讨,结合相关文献和教程内容,提供实用的指导。
HER(Hydrogen Evolution Reaction)是电催化反应中的重要反应,其计算通常涉及吸附能、反应路径、过渡态能量等。VASP通过第一性原理计算(DFT)方法,结合密度泛函理论(DFT)和Kohn-Sham理论,能够精确计算材料的电子结构和反应路径。
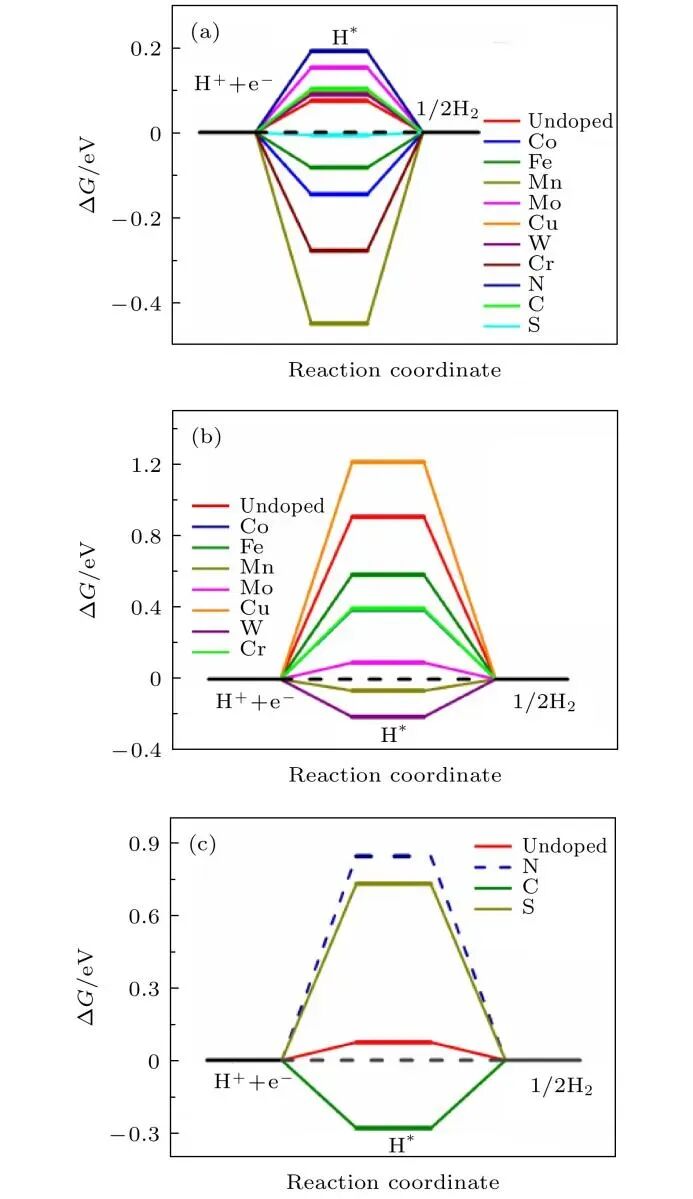
在HER计算中,台阶图(Staircase Diagram)是描述反应路径和能量变化的重要工具,通常包括吸附态、过渡态、产物态等关键点的能量变化。
在VASP计算中,计算效率是关键。VASP支持多种并行计算模式,如MPI(Message Passing Interface)和OpenMP(Open Multi-Processing),通过合理配置并行参数(如NPAR、KPAR、NCORE)可以显著提高计算效率。
例如,设置NPAR为可用处理器数的平方根,KPAR为可用处理器数的平方根,NCORE为每个节点的处理器数,可以有效提升计算速度。
在VASP计算中,精度和收敛性是关键。通过调整计算参数(如K点密度、平面波截断能、混合泛函等)可以控制计算精度。例如,使用GGA(Generalized Gradient Approximation)泛函(如PBE)可以提高计算精度。
此外,通过调整K点密度和混合泛函参数,可以优化计算收敛性,减少计算时间。
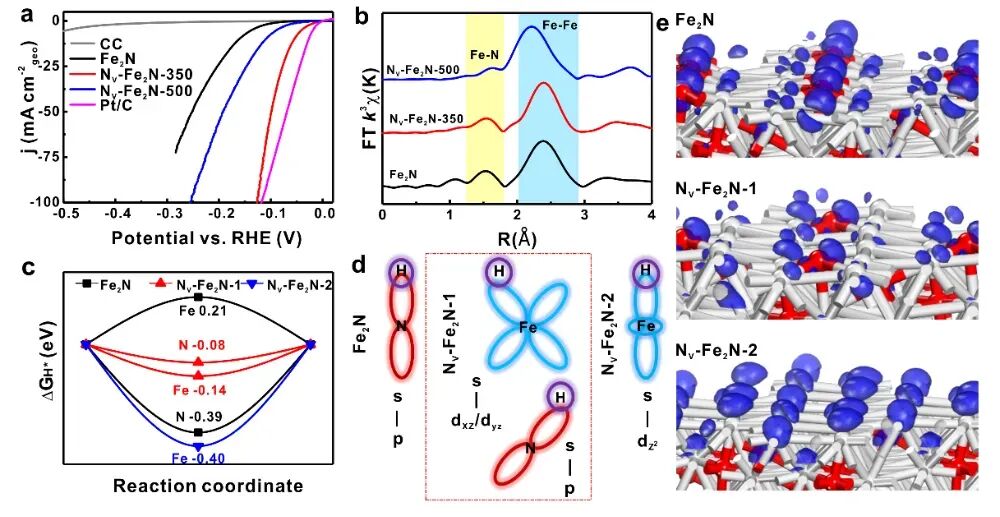
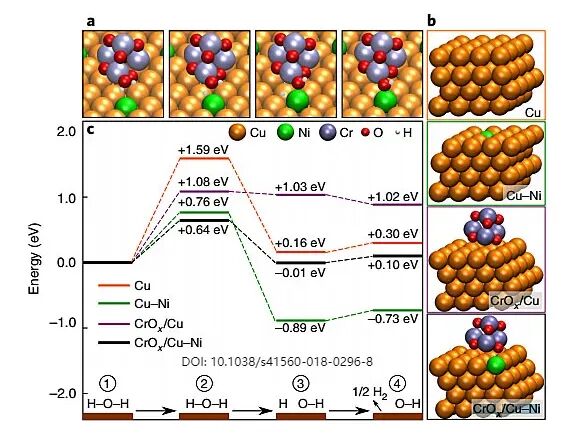
在HER台阶图计算中,过渡态(Transition State)的计算是关键步骤。VASP支持多种过渡态计算方法,如CI-NEB(Constrained Interpolated NEB)和IOPT(Initial Optimization)等。
通过设置合适的参数(如粗算与精算、过渡态搜索算法),可以准确计算反应路径和能量变化。此外,通过差分电荷密度(Differential Charge Density)和d带中心分析,可以进一步理解反应机制。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!