

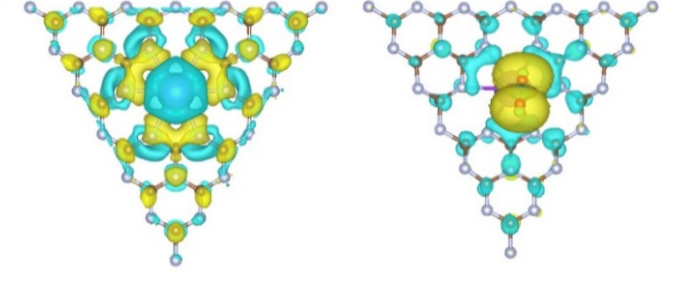
差分电荷密度(Charge Density Difference, CDD)是通过比较体系在相互作用前后电子密度分布而得到的空间函数,能够直观揭示吸附、界面形成、掺杂和缺陷诱导等过程中电子的重新分布及其作用机制。
与基于原子划分的布居分析不同,差分电荷密度以三维等值面的方式在实空间刻画电子积累与耗尽,其典型配色为“黄色/红色表示积累、蓝色表示耗尽”,便于从微观层面识别电荷转移方向、强度与范围。

DOI:10.3866/PKU.WHXB202406005
问题背景
电子在材料中的空间分布及其随相互作用而发生的重排,是决定材料微观结构、能带性质与化学反应性的根本因素。从弱范德华吸附到强共价配位,从二维异质结到金属/半导体界面,电子在纳米尺度的迁移、极化与杂化都会在可观测层面改写吸附能、界面偶极、载流子输运与催化活性等关键指标。
密度泛函理论(DFT)作为主流的第一性原理工具,能够给出高分辨率的电荷密度,在此基础上构建的差分电荷密度方法以“相互作用前后之差”的思想,直观地把复杂的量子多体效应转换为可视化的空间图像,从而为研究者提供了追踪电荷转移路径与判读轨道杂化强弱的窗口。
尽管 Mulliken、Hirshfeld 或 Bader 等布居分析能以原子为单位给出电荷获得与损失的数值,但它们不可避免地依赖划分准则而存在方法学差异;与之互补的差分电荷密度避免了“把电子分给谁”的人为裁决,而以等值面或切片热图的方式揭示电子在实空间的连续分布变化,因而尤其适合分析局域化与非局域化效应并存的界面体系。
在吸附、掺杂和缺陷工程等问题上,差分电荷密度能将“电子从何处来、到何处去”的微观图景清晰呈现,进而与能谱信息结合,建立“电荷转移—配位化学—反应能垒/吸附强度”的因果链条。
DOI:10.1016/j.cej.2025.163923
定义与理论基础
差分电荷密度Δρ(r) 的定义可写为:Δρ(r)=ρ_total(r)−∑_i ρ_i(r),其中 ρ_total(r) 为包含相互作用的完整体系电荷密度,ρ_i(r) 为在相同超胞、赝势与数值参数下、保持原子在同一空间位置的各个组分 i 的孤立电荷密度。
这种“同构同参”的构造确保了差分运算具有物理一致性与可比性,避免了由几何与数值差异诱发的伪影。在可视化上,人们通常以黄色或红色标示电子积累区域(Δρ>0),以蓝色标示电子耗尽区域(Δρ
值得强调的是,Δρ并不等价于“净电荷”,而是相互作用导致的“局域再分布”,其积分在整个超胞上等于零,局部积分则反映给定区域的电荷流入或流出。
从微观机制看,差分电荷密度的起源可追溯到波函数重叠与费米能级对齐导致的轨道杂化以及静电势重整。
当吸附分子靠近金属或半导体表面时,其前线轨道(如π、π* 或孤对电子)与基底的 d、p 态发生混成,伴随能级的分裂与填充变化,从而在实空间产生可观测的电子积累/耗尽图样;在极化基底或带有缺陷电偶极的界面,则可能出现显著的电荷推挤与屏蔽效应,形成指向界面的电荷双层。
因此,差分电荷密度不仅是图像化的“后处理”,更是量子化学相互作用在实空间的直接投影,与态密度、能带与静电势剖面共同构成理解界面相互作用的多视角证据链。
计算流程与可视化参数
可靠的差分电荷密度依赖严格的一致性流程。第一,构建包含所有相互作用的完整模型并充分弛豫,得到自洽的ρ_total(r);第二,在完全相同的超胞、截断能、k 点、赝势与网格设置下,将各组分在相同原子坐标处“冻结”计算孤立态电荷密度 ρ_i(r),避免几何差异引入非物理信号;第三,使用后处理程序(如 VASP 的 chgsum.pl/chgdiff、Quantum ESPRESSO 的 pp.x 或自编脚本)进行网格对齐与差分。
可视化时,等值面阈值需结合体系调节:过低会淹没噪声,过高又会遗漏弱相互作用,对金属/分子吸附与二维异质结而言,0.001–0.005 e/ų 常见且可比。
图像解读的稳健性离不开切片厚度、平滑方式与色标范围的科学选择。对于层状体系,沿法向的二维切片能揭示层间电荷双层及偶极方向;对于孤立活性位,三维等值面更能呈现“环绕式”的积累与耗尽外形。
必要时应给出多阈值并列图,以展示从“强耦合核心”到“弱极化外延”的层级结构;若研究目标涉及定量比较(如不同吸附构型、不同金属位),推荐在相同色标与阈值下制图,以避免视觉误导。
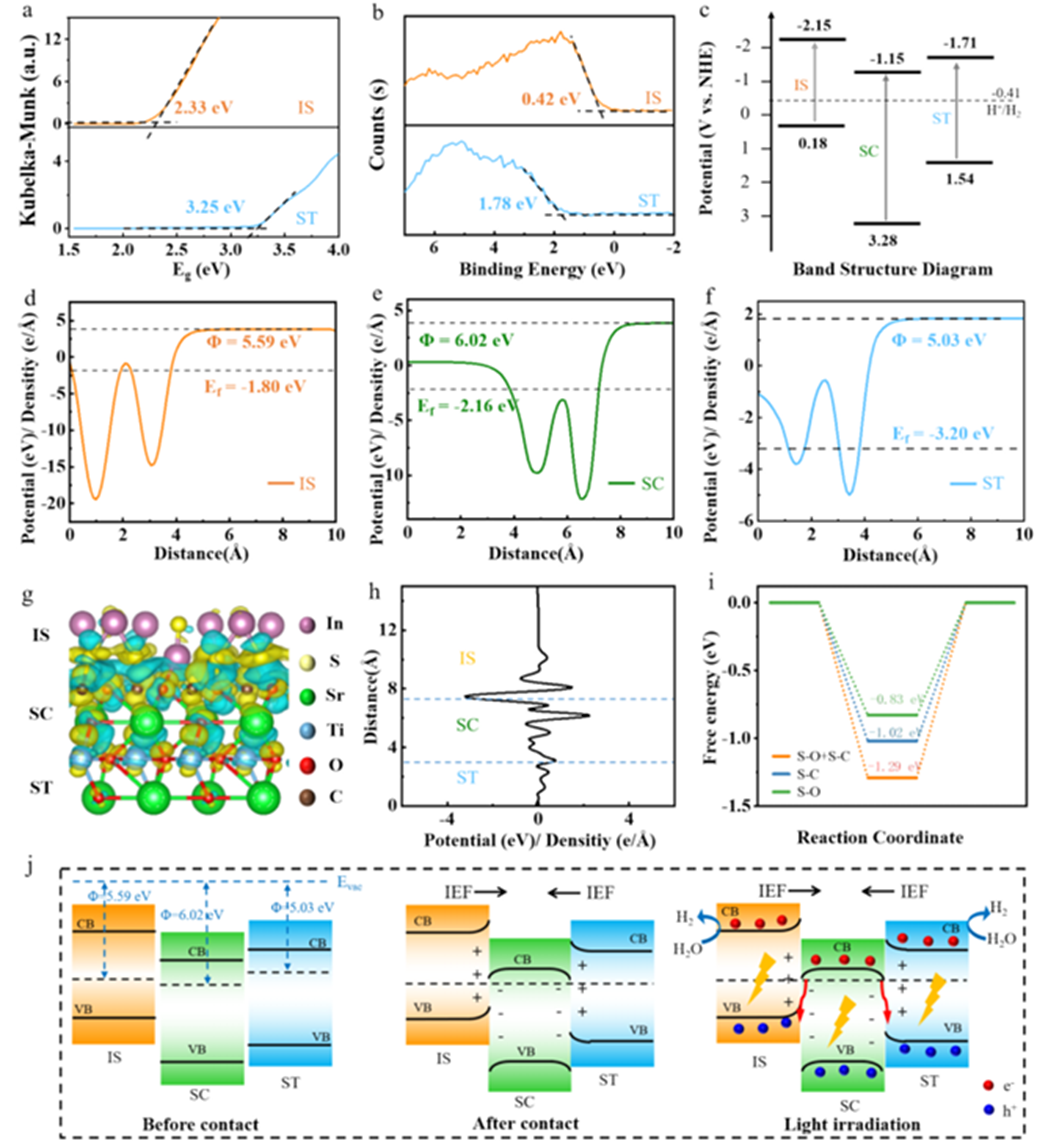
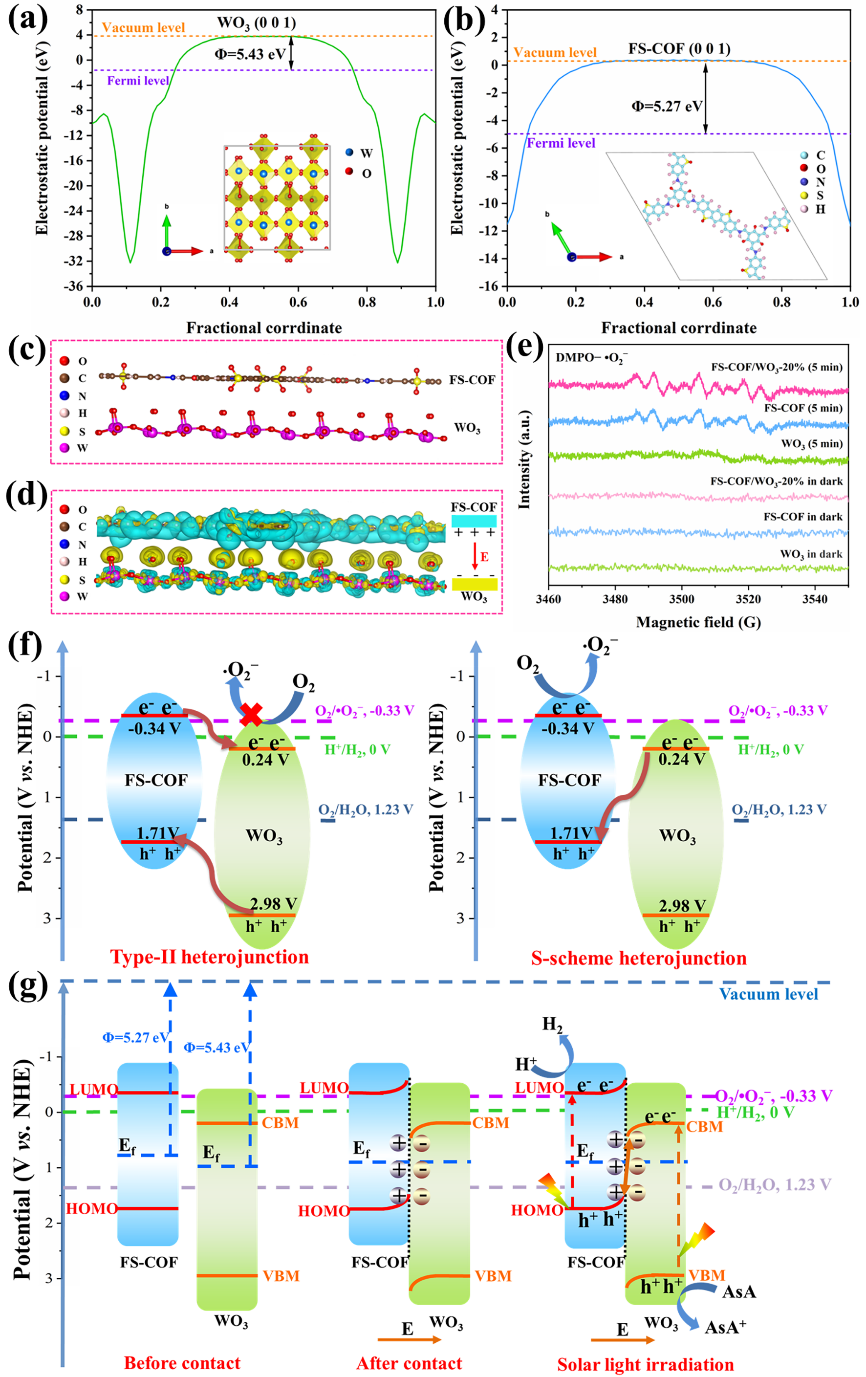
此外,将 Δρ与平面平均电荷密度、沿法向积分的累积电荷曲线或静电势差ΔV(z) 并置,可进一步量化界面偶极强度与电场方向,为能带弯曲与功函数变化提供佐证。
分析方式与多源证据整合
差分电荷密度的核心价值在于与能谱和布居的交叉印证。首先,将Δρ的局域特征与投影态密度(PDOS)相互映照:若在吸附后观察到分子 π* 轨道与基底 d 态在费米能级附近出现杂化峰,并伴随 Δρ在分子反键轨道区域显著积累,即可推断电荷向分子转移并形成弱配位键;若Δρ显示基底表面原子周围耗尽而分子孤对电子方向积累,则提示路易斯酸-碱配位增强。
其次,结合能带结构与功函数变化判断界面偶极与费米能级对齐的结果,例如 p 型基底上电子向分子转移会抬升功函数并导致价带顶下移。
在定量层面,Bader 电荷提供了以原子为单位的电荷转移数值,与 Δρ的空间分布互为补充。常见做法是在Δρ显著区域定义一个积分体(如围绕活性位的 Voronoi 或手动盒子),对 Δρ体积分以估计“局部电荷变化”,并与 Bader 电荷差 Δq 进行对比以验证稳健性。
此外,电子局域函数(ELF)可以揭示键合类型的改变:若 Δρ显示金属-杂原子之间积累,且 ELF 在键中点由低值升高,说明共价性增强;若 Δρ呈现较远程的面外积累而 ELF 变化有限,则更可能是静电极化或金属态屏蔽。
通过“Δρ—PDOS—Bader—ELF—功函数/能带”的组合拳,可将相互作用的几何—电子—能量三域信息闭环。
典型应用
在催化领域,差分电荷密度已成为甄别活性位与解释反应能垒变化的标配工具。对单原子催化剂而言,载体向金属原子的电子馈赠会改变其 d 态占据,从而调节 d 带中心并影响 *H、*O、*OOH 等中间体吸附强度;Δρ中在金属位四周的“环形积累”常指示电子富集与较弱的质子耦合,而在载体相邻阴离子位的耗尽则暗示配位环境的再平衡。
就金属簇或界面合金而言,Δρ能揭示配位不饱和位的强化活性来源——电子向低配位位点聚集使其成为亲反应中间体的“接收端”。
DOI:10.1016/S1872-2067(25)64664-X
在电极/电解质界面中,差分电荷密度刻画的第一层溶剂与离子鞘层电荷重排,直接关联 SEI 成膜途径与过电位。若碳酸酯分子的羰基区出现显著积累而电极表面出现耗尽,说明发生了向分子 LUMO 的电子注入,解释其优先生成还原产物;对于固态电解质与金属锂界面,Δρ的双层分布与电势梯度可用于预测离子迁移的首通道与界面稳定性。
在二维异质结与范德华堆叠体系,层间Δρ呈现波纹状的交替积累/耗尽,配合功函数差判断层间电荷转移方向,进一步联系到激子束缚与带隙重整,为光电器件设计提供依据。
实操指南与常见误区
为了获得可复现且可比较的Δρ结果,必须遵循“几何对齐、参数同一、网格等距”的三大原则。几何对齐要求孤立组分与复合体系采用完全相同的原子坐标(含真空层与约束),避免弛豫差异带来伪信号;参数同一要求使用相同的赝势、平面波截断、k 点采样与电子温度;网格等距则确保不同计算的电荷密度栅格可逐点相减。
在 VASP 中可通过保留 CHGCAR、CHG 文件并使用脚本进行差分;在 Quantum ESPRESSO 中可用 pp.x 输出并用后处理对齐。如果研究对象包含强相关电子或自旋极化,建议统一加入 U 或考虑非共线自旋,并在可视化时分别展示自旋分量的 Δρ。
等值面的选择是最常见的误区来源。过度追求“好看”的高阈值会导致对弱耦合或长程极化的忽视,而过低阈值又可能把数值噪声当作物理信号。经验上可在 0.001、0.002、0.005 e/ų 三档并列给图,并固定色标范围以便横向比较。
同时应避免把 Δρ的视觉强弱直接等同于反应能量差,因为Δρ反映的是空间再分布而非总能变化;正确做法是将Δρ的局域特征与 PDOS、Bader 电荷与吸附能变化建立关联,在多源证据一致时给出稳健结论。必要时给出平面平均 Δρ(z) 与功函数变化以支持界面偶极的论断。
局限性、可比性与定量化趋势
差分电荷密度的局限性首先体现在其半定量属性:不同体系、不同阈值与色标下的图像很难作绝对意义的强弱比较;其次,Δρ的可视化会受到超胞尺寸与真空厚度的影响,过薄真空可能导致层间耦合伪影,过小超胞则易引发镜像相互作用。
此外,强关联或金属态体系中费米展宽与自洽收敛精度也会显著影响Δρ的细节。因此,跨研究工作的对比必须在相同参数与后处理协议下进行,并尽可能提供原始电荷密度数据以便复现。
推动Δρ定量化的一个方向是与可积体定义、Bader 体积分与机器学习描述符结合。通过在标准化阈值下提取“积累/耗尽体积、体积分、形状矩”(如球谐矩或惯性张量),可把图像转化为可比较的向量表征,再与吸附能、反应能垒或电催化指标进行回归关联,从而建立“Δρ—性质”的可转移关系式。
随着公共数据集与工作流程平台的成熟,差分电荷密度有望成为高通量筛选中的一类一阶判据,辅助快速锁定电荷转移受控的优异材料组合。
总结
差分电荷密度以简洁的差分构造与直观的空间表征,连接了量子化学相互作用与可视化解读之间的鸿沟。它既能在单原子催化、界面工程与二维异质结中揭示关键的电子转移路径与轨道杂化模式,又能在电极/电解质界面与缺陷工程中刻画偶极与极化的层级结构。
只有在严格的一致性计算与多源证据校验下,差分电荷密度才能发挥从定性洞察走向半定量描述的潜力,并与高通量与数据驱动范式融合,成为新材料发现与机理解析的“第一现场证据”。展望未来,围绕 Δρ的标准化、定量化与开放化将持续提升其学术与工程价值。