

在固体物理与材料科学研究中,能带(band structure)是描述晶体中电子行为的核心物理量之一。通过分析材料中电子的允许能级与禁止能级区间,我们能够深入理解其导电性、光学响应、磁性以及热电性能等一系列关键性质。
能带结构不仅揭示了电子如何在周期性势场中传播,还奠定了我们对金属、半导体与绝缘体区别的理论基础。在现代计算材料学中,通过第一性原理计算所获得的能带图谱,已成为预测与解释实验现象的重要工具。
能带的研究贯穿材料设计、器件工程乃至纳米科技等多个层面。从电子跃迁所需能量的精确计算,到异质结界面态的调控,再到新型二维材料与拓扑绝缘体的能带奇异性研究,能带分析均发挥着不可替代的作用。
因此,系统理解能带的基本概念、理论来源、建模方法以及实际解读方式,对于任何从事材料模拟、半导体开发或凝聚态理论研究的科研人员而言,都是基础而又关键的知识储备。
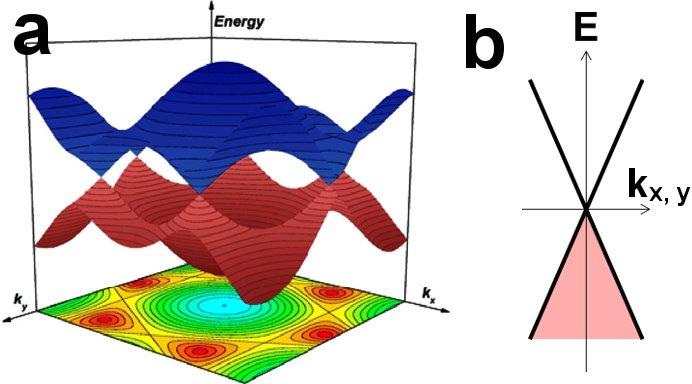

能带的起源与理论模型构建
能带的概念来源于电子在周期性势场中的行为模式。根据布洛赫定理,处于晶体周期性势能下的电子,其波函数可以写成平面波与周期函数的乘积形式。
在自由原子中,电子能级是离散的;但在晶体中,原子之间相互作用导致这些能级展宽为带状结构,从而形成所谓的“能带”。
具体而言,当大量原子靠近形成晶体时,原子轨道间发生重叠,形成能量连续的带,并在某些能量区域中出现禁止态,称为禁带(band gap)。
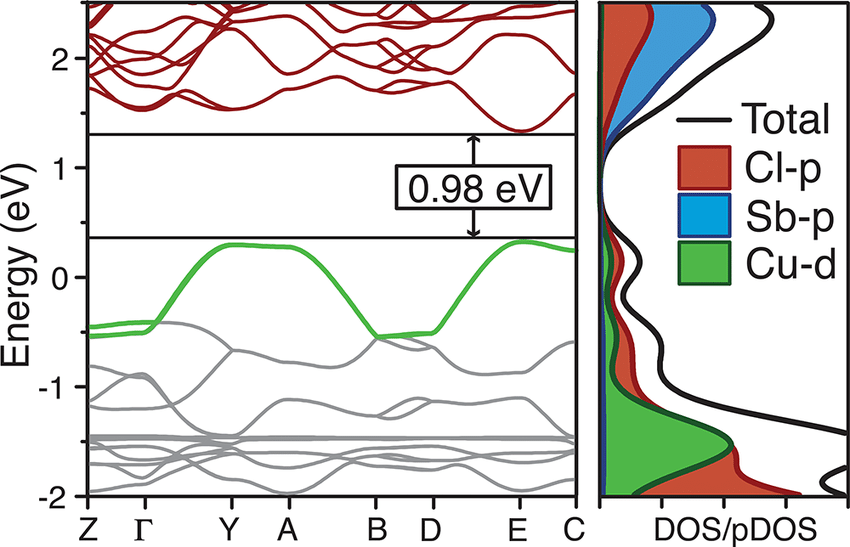
DOI:10.1021/jacs.7b04119
不同材料中原子种类、晶体结构与键合方式不同,所形成的能带特征也截然不同。金属的能带呈现部分填充态,使其具备良好导电性;而半导体和绝缘体则有明显的能带间隙,且价带顶(VBM)与导带底(CBM)决定其激发行为。
在能带形成模型中,紧束缚近似(tight-binding)与几乎自由电子模型(nearly free electron model)是两种典型理论方法,分别适用于电子局域化与离域化程度不同的体系。前者强调原子轨道耦合,后者则关注布拉格反射与能隙打开的物理机制。
能带计算方法
现代计算材料科学通常采用密度泛函理论(DFT)作为能带计算的基础框架。在DFT中,体系总能量以电子密度为自变量,通过求解Kohn-Sham方程获得体系的本征态与本征能,进而构造能带结构图谱。
以平面波为基组的计算方法,如VASP、Quantum ESPRESSO与ABINIT,是最常用于晶体材料能带分析的软件平台,它们可高效处理周期性势场下的能量本征值问题。
在能带结构计算中,k点采样方案对于图像精度具有重要影响。由于能带图仅显示晶体的高对称路径,k空间路径的选取(如Γ–X–M–Γ等)需结合晶体的点群对称性与布里渊区形状进行优化。
此外,为获得更真实的带隙值,常需引入杂化泛函(如HSE06)或准粒子GW方法对传统GGA或LDA结果进行修正,避免低估导带位置。尤其在对光学带隙、激发态谱图或拓扑特征进行高精度分析时,这些方法的使用变得不可或缺。
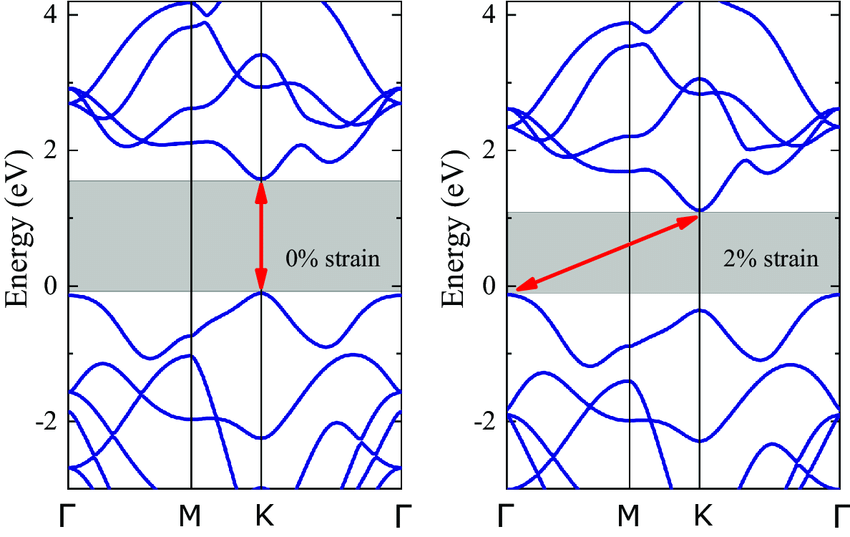
DOI:10.1038/s41699-019-0123-5
能带结构图谱的解读方式
能带结构图通常以k点路径为横坐标,电子能量为纵坐标,表示电子在不同晶体动量状态下的能量分布。通过分析能带图,可以判定材料的导电性、带隙类型(直接或间接)、电子有效质量等重要性质。
直接带隙表示价带顶与导带底出现在同一k点,有利于光生电子-空穴对直接复合,适合光电器件;间接带隙则需借助声子辅助跃迁,效率较低。
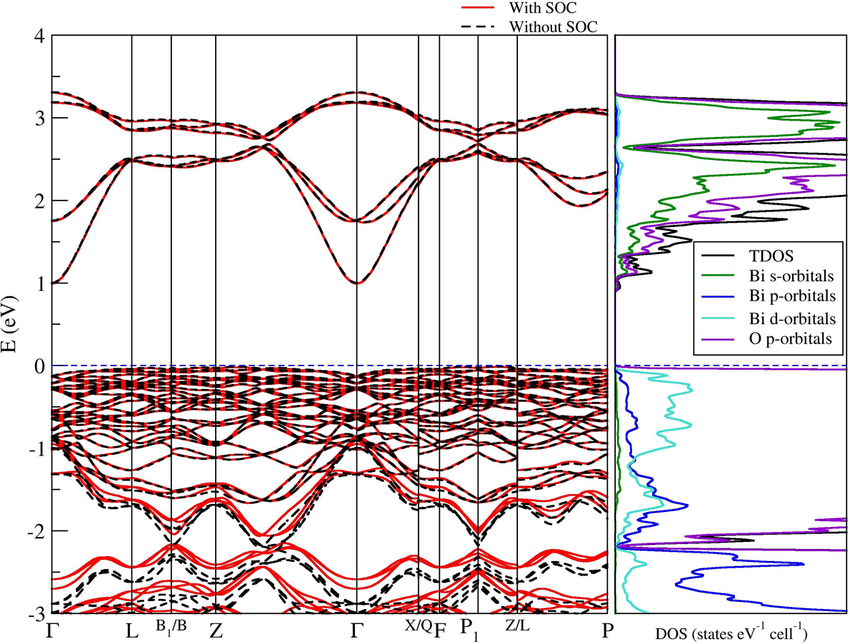
DOI:10.1103/PhysRevMaterials.4.075401
此外,通过投影态密度(Projected DOS, PDOS)或轨道成分分析,可以明确不同原子或轨道类型对能带的贡献。例如,过渡金属氧化物中常见导带主要来源于金属3d轨道,而价带由氧2p轨道主导。
这种成分信息对于理解杂化机制、判断电子跃迁方向、构建有效电子哈密顿量至关重要。在半导体异质结设计中,能带对齐(band alignment)分析更是预测电子流动与界面性能的核心依据。
案例分析
以硅为例,其为典型的间接带隙半导体,导带底出现在X点,价带顶位于Γ点。其能带结构显示出典型sp³杂化轨道特征,价带展宽明显,导带较为平坦,电子有效质量较大,这一结构特征解释了硅在太阳能电池中的使用效率相对有限。
而砷化镓(GaAs)则为直接带隙材料,带隙较小,电子迁移率高,是发光二极管与激光器的核心材料之一。
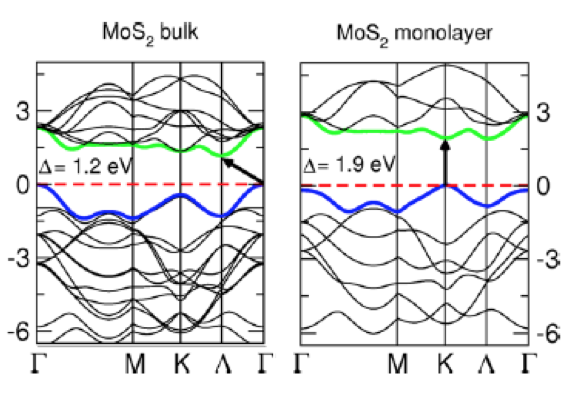
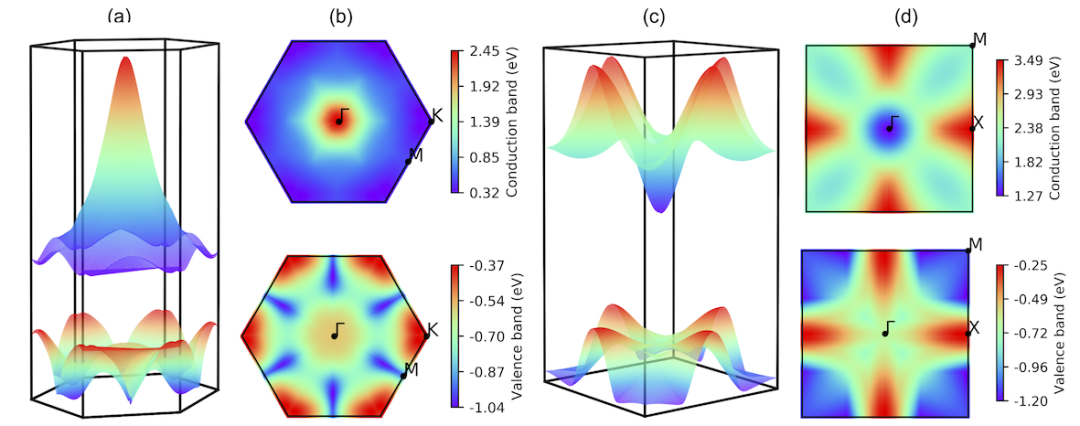
在新兴二维材料如MoS₂中,其能带在单层时呈现直接带隙,而在多层堆叠后转变为间接带隙,这种维度调控效应赋予材料在纳米电子器件中的应用潜力。
类似地,拓扑绝缘体如Bi₂Se₃具有倒置能带结构,在表面形成受保护的无质量Dirac态,是研究自旋电子学与量子输运的热点方向。通过这些案例我们可以看出,不同结构与成分对能带的调控能力已成为材料性能优化的基本路径。
能带与功能性质之间的联系
能带结构不仅决定了材料的基本电学行为,还与光学吸收、热输运、超导转变、磁性等多种性质密切相关。
例如在热电材料中,带隙大小与电子有效质量的控制对Seebeck系数与功率因子的优化至关重要;而在光催化材料中,带隙宽度与导带位置决定了其是否具有还原水的能力。
在磁性材料中,能带的自旋分裂决定了其铁磁性强度与自旋极化率,而半金属材料中,能带结构呈现一自旋通道导电、另一自旋通道绝缘的态密度特征,是发展自旋注入器的理想材料。
对这些功能性应用的深入探索,要求研究者能够从能带结构出发,提取关键的电子结构指标,结合态密度、费米面、布里渊区拓扑等手段进行系统性分析。
从能带到材料设计的理论指导
现代材料设计强调从“电子结构出发”进行功能性预测与性能筛选。在大规模高通量计算平台(如Materials Project, AFLOW, OQMD)中,能带结构与带隙数据成为分类与评估材料性能的基础指标。
通过统计不同晶系、原子构型与价层填充对能带的影响,可以建立起以能带为中心的机器学习模型,实现新材料的快速发现与性能预测。
更进一步,结合能带工程策略如应力调控、外场作用、掺杂替换等手段,研究者可以实现对能带宽度、带隙类型、载流子浓度的精准控制,从而构建出面向特定应用的材料体系。
例如在二维电子气、量子霍尔效应、热电器件等领域,能带结构的细致调控构成其物理行为与器件性能提升的核心动力。因此,能带不仅是表征材料的“光谱图”,更是设计新材料的“地图”。
总结
综上所述,能带作为描述电子在晶体中运动行为的核心图谱,贯穿材料科学的多个分支。
它揭示了电子能态的排列方式,确定了材料的导电性质、光学响应与热力行为,并通过第一性原理计算成为现代材料研究中不可或缺的工具。能带的计算、解读与工程调控已成为推动新材料发现与器件创新的关键路径。
随着计算能力的提升与理论工具的完善,能带分析将更趋精细与多维,涵盖自旋轨道耦合、拓扑特征、强关联行为等复杂电子态。同时,能带图谱也将更多地融入数据驱动材料发现流程,与实验数据、表面态特征、缺陷态分布形成协同分析网络。
未来,能带结构将不仅用于“解释”,更将用于“预测”与“引导”,成为连接基础物理与应用设计的桥梁。