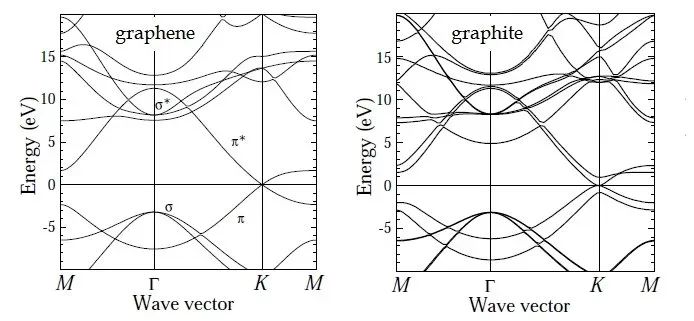
能带结构(band structure)是凝聚态物理、材料科学和量子力学中的一个重要概念,用于描述系统中能量间隔内的微观状态数。它在计算材料的电子结构、光学性质、催化性质等方面具有重要意义。华算科技朱老师将详细介绍二维材料石墨烯能带结构计算步骤和数据处理方法。
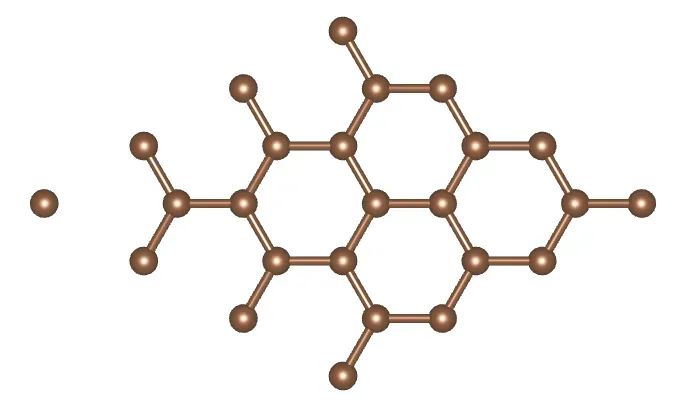
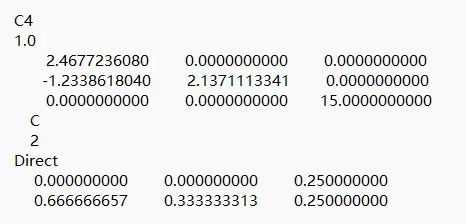
首先通过晶体结构数据库下载层状材料石墨的cif文件,并生成POSCAR文件,然后去掉一层C原子,获得石墨烯结构,如下图所示。
ICHARG=2 #从原子电荷密度产生体系初始电荷密度
EDIFF=1E-5 #电子波函数能量收敛标准1E-5 eV
IBRION=2 #共轭梯度法优化晶体结构和原子坐标
NSW=100 #晶体结构和原子坐标优化步数最大100步
EDIFFG=-0.1 #原子残余力小于0.1 eV/A
ISMEAR=0 #费米能级附近电子占据数为高斯分布
Automatic generation #注释行
完成结构优化计算后,保持KPOINTS,POTCAR文件不变,将CONTCAR文件复制成POSCAR文件,并对结构优化的INCAR文件作如下修改:
完成自洽计算后,保持POTCAR,POSCAR文件不变,将自洽计算的CHGCAR,WAVECAR保留下来,并对自洽计算的INCAR和KPOINTS文件作如下修改:
k-points along high symmetry lines #注释行
0.333333 0.333333 0 #高对称性K点2
0.333333 0.333333 0 #高对称性K点2
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!