双原子催化剂概括
双原子催化剂(Dual – Atom Catalysts,DACs)是一类具有独特结构和性能的新型催化剂,其核心特征是在催化剂表面或内部存在两个相邻的活性金属原子,这两个原子可以是同种金属(如 Cu – Cu),也可以是不同金属(如 Cu – Fe) 。
与传统的纳米颗粒催化剂和单原子催化剂相比,双原子催化剂具有以下显著的结构特点:

双活性位点:双原子催化剂的双活性位点设计是其区别于其他催化剂的关键特征之一。这两个相邻的活性位点能够提供协同作用,使得催化剂对反应物的吸附和活化能力得到显著增强。
例如,在某些加氢反应中,一个金属原子可以优先吸附氢气分子并将其解离为氢原子,而另一个金属原子则负责吸附反应物分子,促进氢原子与反应物分子之间的反应,从而提高反应速率和选择性。
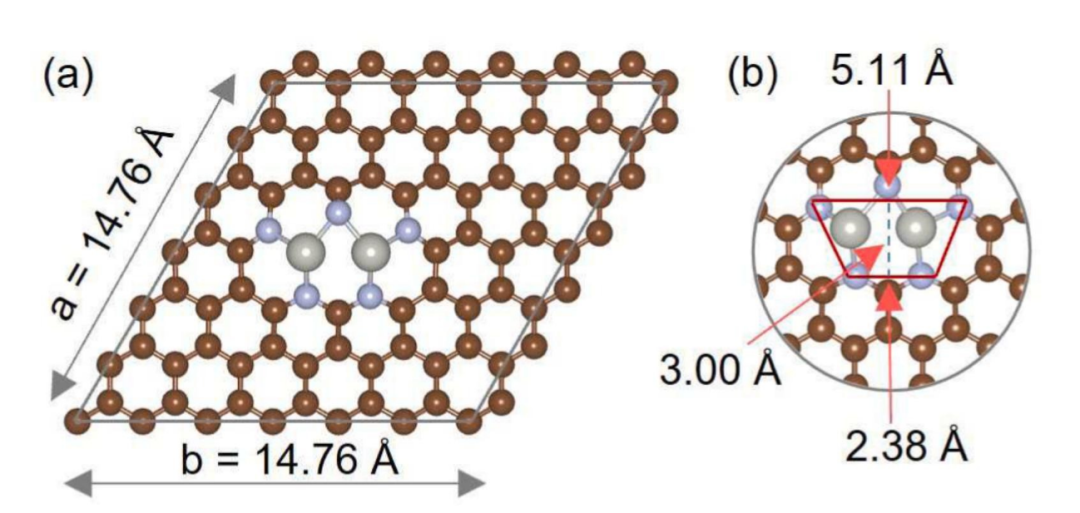
精确的原子间距:双原子之间的距离通常在 0.2 到 0.5nm 之间,这种精确控制的原子间距对于优化反应物的吸附和中间体的形成起着至关重要的作用。
合适的原子间距能够使两个活性位点之间产生有效的电子相互作用和空间协同效应,促进反应物分子在两个位点之间的协同吸附和转化,从而提高催化效率。
例如,在一些电催化反应中,精确的原子间距可以调节反应中间体在两个活性位点上的吸附能和反应路径,降低反应的过电位,提高反应的动力学性能。
高度分散:双原子催化剂中的金属原子高度分散在载体表面或内部,避免了金属纳米颗粒的团聚现象。这种高度分散的结构使得金属原子的活性位点能够得到充分利用,极大地提高了金属原子的利用率。
与传统纳米颗粒催化剂相比,双原子催化剂在相同金属负载量下具有更多的活性位点,从而能够表现出更高的催化活性。
例如,在一些贵金属催化剂中,高度分散的双原子结构可以在减少贵金属用量的同时,保持甚至提高催化剂的性能,降低催化剂的成本。
双原子位点协同效应的作用机制
双原子位点的协同效应是其展现优异催化性能的核心,其作用机制主要涵盖协同吸附与活化、电子转移与调节以及中间体稳定化这几个关键方面。
这些机制并非孤立存在,而是相互关联、协同作用,共同推动催化反应朝着高效、高选择性的方向进行。深入探究这些作用机制,对于理解双原子催化剂的工作原理以及开发高性能的催化剂具有至关重要的意义。
协同吸附与活化
在催化反应这一复杂且关键的化学过程中,反应物分子在催化剂表面所发生的吸附与活化,堪称整个反应得以顺利开启的起始关键步骤。这一初始步骤的效率犹如反应的“命门”,直接且紧密地关联着整个反应的速率快慢以及活性高低。
双原子位点,作为催化剂领域中独具魅力的存在,凭借其与众不同、独一无二的结构特征以及特殊的电子特性,展现出令人瞩目的优势。
它能够针对反应物分子巧妙地发挥协同吸附与活化作用,通过这种协同效应,打破反应物分子原有的稳定状态,促使分子内化学键的重新排列与组合,进而显著地提升这一关乎全局的关键步骤的效率,为高效的催化反应奠定坚实基础。
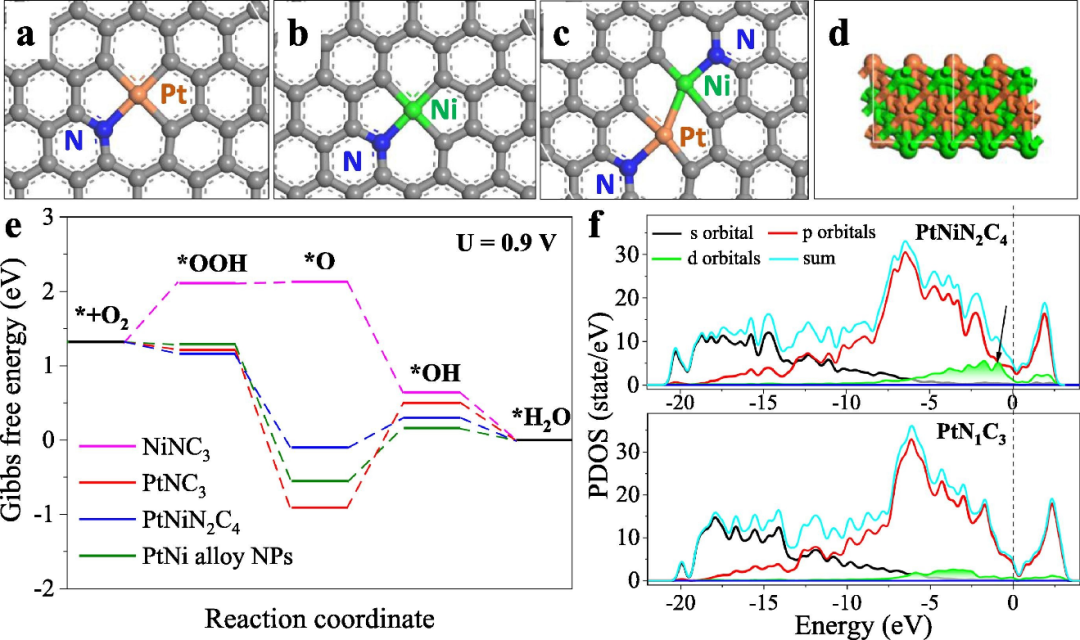
电转移与调节
双原子位点之间的电子转移是协同效应的另一个极为关键且不容忽视的重要作用机制。在各类催化反应体系中,这种电子转移过程扮演着举足轻重的角色,它能够对反应物的吸附强度展开极为精细且精准的调节。
具体而言,通过微妙地改变原子间的相互作用,使得反应物在催化剂表面的吸附既不会过强而导致反应难以进行后续步骤,也不会过弱而无法有效引发反应,从而恰到好处地优化整个反应路径。
如此一来,便能够显著地提高催化反应的效率,在更短的时间内获得更多的目标产物,同时还能大幅提升反应的选择性,使反应朝着生成期望产物的方向高效进行,避免产生大量不必要的副产物。
在双原子催化剂里,两个紧密相邻的金属原子,由于它们各自电负性的不同,即对电子吸引能力存在差异,以及电子云密度在空间分布上的特性有所区别,这些电子结构方面的显著差异会促使在原子间形成一条独特而特定的电子转移路径,并且产生独特的电荷分布状态。
这种电子转移现象,一方面直接影响了金属原子自身原本的电子状态,使其处于一种更为活跃或者更为稳定的电子构型;另一方面,它还会通过改变催化剂表面的电荷密度,让催化剂表面某些区域的电荷富集或者贫化,以及改变电子云在空间的分布形态,进而对反应物分子在催化剂表面的吸附行为和反应活性产生十分显著且深远的影响,从微观层面上左右着整个催化反应的进程和最终结果。
中间体稳定化
在催化反应过程中,反应中间体的稳定性对反应的选择性和活性起着至关重要的作用。从微观层面来看,不稳定的中间体内部化学键的结合力相对较弱,在反应体系中面临各种分子的碰撞以及能量的起伏变化时,其结构极易发生改变,进而容易引发一系列副反应。
这些副反应会消耗反应物,使更多的原料偏离生成目标产物的反应路径,最终导致目标产物的选择性降低。而稳定的中间体则截然不同,其原子间的相互作用使得结构紧凑且有序,在复杂的反应环境中能保持相对稳定。
这种稳定性能够促使反应体系中的能量分布更加合理,让反应沿着期望的路径进行,就像为反应指明了方向,避免了能量的无效损耗和不必要的反应步骤,进而提高目标产物的生成效率。
双原子位点凭借其独特的电子结构和空间构型,能够通过与反应中间体形成特定的相互作用,比如通过配位键、氢键等弱相互作用,稳定关键中间体。
这种稳定作用能够有效调控反应中间体的电子云分布,优化其反应活性位点,从而极大地提高反应的选择性,使得反应朝着生成目标产物的方向高效进行。
案例分析
双原子协同催化:CO₂加氢反应中的能垒调控与路径选择
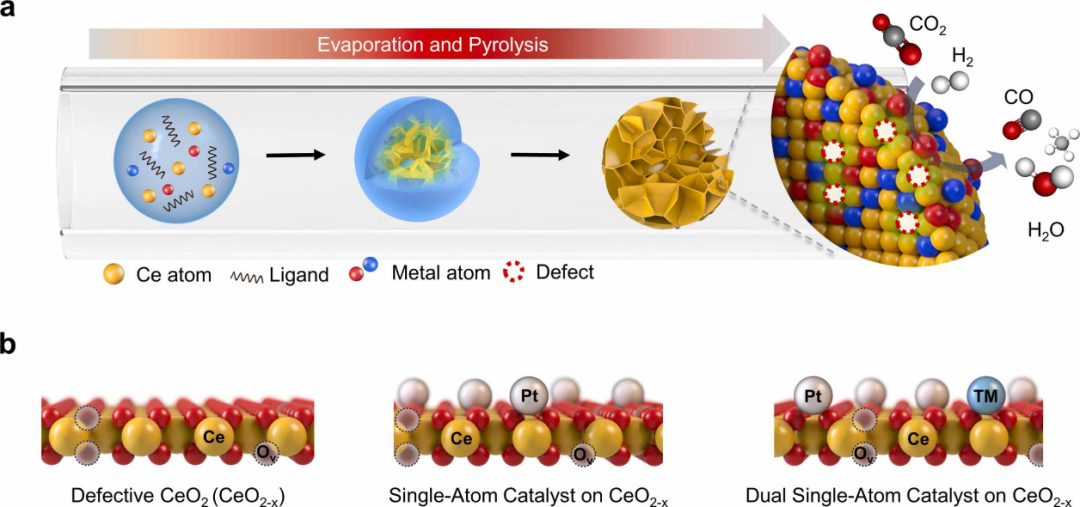
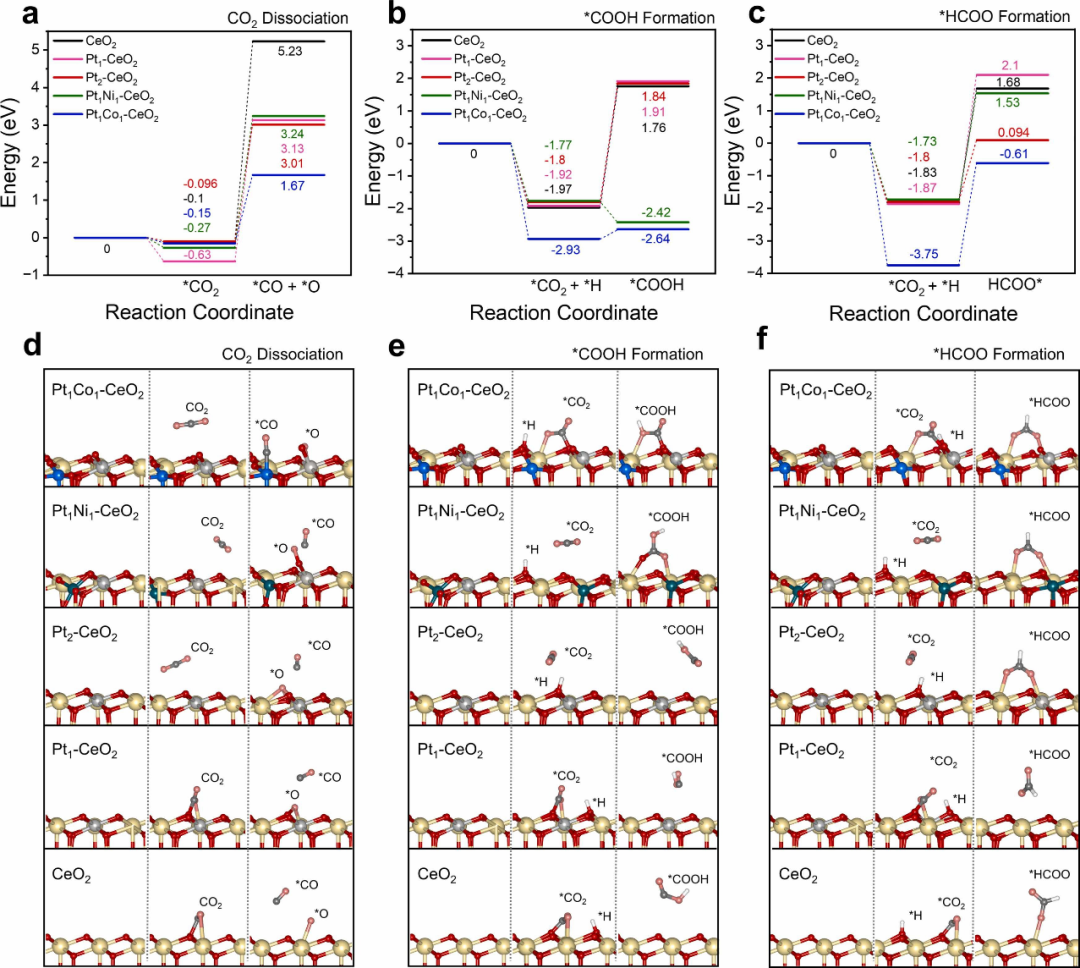
下图通过密度泛函理论(DFT)计算深入揭示了双位点催化剂(Pt₁Co₁-CeO₂和Pt₁Ni₁-CeO₂)在CO₂加氢反应中的显著优势,其核心在于双金属协同效应与载体缺陷的协同调控,从而在反应活性、选择性和稳定性方面展现出超越单一位点催化剂的卓越性能。
从反应机理来看,双位点催化剂通过优化金属–金属(Pt-Co/Ni)与金属–载体(Pt-CeO₂)的电子相互作用,显著降低了CO₂活化关键步骤的能垒。
在CO₂直接解离路径中,Pt₁Co₁-CeO₂的反应能垒(2.30 eV)比单一位点Pt₁-CeO₂(3.75 eV)降低了近40%,这种能垒的降低源于Co原子对Pt电子结构的调控,使得Pt位点对CO₂的吸附构型更有利于C=O键的断裂。
与此同时,Pt₁Ni₁-CeO₂在羧基路径(*COOH形成)中表现出放热特性(-0.65 eV),这一现象揭示了Ni原子通过促进H原子向CO₂的转移,显著加速了*COOH中间体的生成,为后续CH₄的形成提供了关键前驱体。
这种路径选择性的差异完美解释了实验观察结果:Pt₁Co₁-CeO₂在200-400°C范围内保持98%以上的CO选择性,而Pt₁Ni₁-CeO₂则表现出更高的CH₄产率。
进一步分析发现,双位点催化剂的优势不仅体现在能垒降低,更在于其对反应中间体吸附强度的精准调控。Pt₁Co₁-CeO₂对*HCOO中间体的强吸附(-0.61 eV)使其能够稳定该物种并促进其分解为CO,而不会过度加氢生成CH₄;相比之下,Pt₁Ni₁-CeO₂对*COOH的稳定作用则引导反应向CH₄路径发展。
这种“分子水平的设计“得益于双金属位点的电子互补效应:Co的3d电子与Pt的5d轨道杂化形成了新的电子态,在费米能级附近产生了更高的电子密度,这既增强了CO₂的活化能力,又通过调节氧空位浓度优化了H₂的解离吸附。
实验表征数据为此提供了有力佐证:H₂-TPR显示Pt₁Co₁-CeO₂在150-200°C区间具有更高的H₂消耗量,表明其表面晶格氧物种更易被活化;而CO-DRIFTS则观察到Pt₁Co₁-CeO₂中孤立Pt单原子的特征峰增强,证实了Co原子对Pt分散性的促进作用。
从催化剂稳定性角度,双位点结构通过形成Pt-M-O-Ce(M=Co/Ni)的强相互作用网络,有效抑制了金属原子的迁移和团聚。从理论层面深入分析,双位点协同效应的物理本质在于d带中心的调控。
DFT计算显示,Pt₁Co₁-CeO₂中Pt的d带中心相对于费米能级上移了0.3 eV,这增强了其与CO₂反键轨道的重叠,从而促进电荷转移;同时Co的引入在CeO₂(111)表面形成了特定的氧空位构型,这些空位作为电子陷阱稳定了反应中间体。
这种原子尺度的“双活性位点“设计——Pt位点主导H₂活化,Co/Ni修饰的氧空位主导CO₂吸附——实现了反应物分子的分区活化,避免了竞争吸附导致的活性位点阻塞。
对比传统合金催化剂,双单原子构型确保了每个金属原子都处于最大暴露状态,使原子利用率接近100%,这解释了为何Pt₁Co₁-CeO₂的质量活性是Pt/CeO₂纳米颗粒的8倍以上。
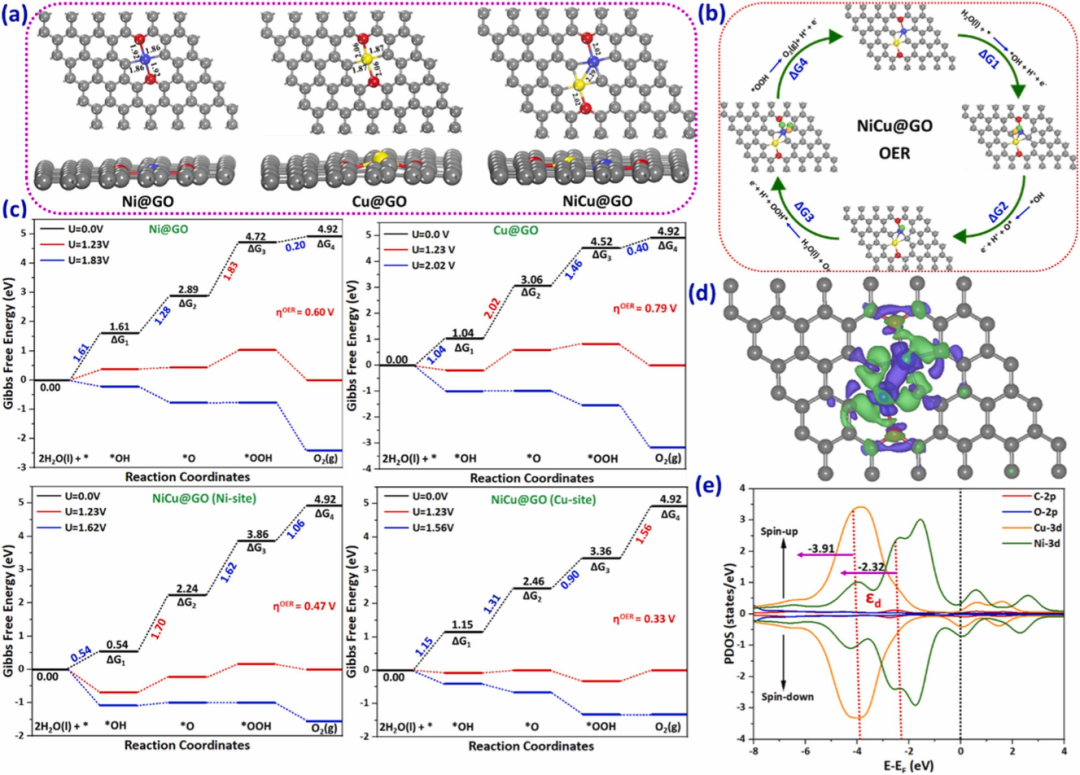
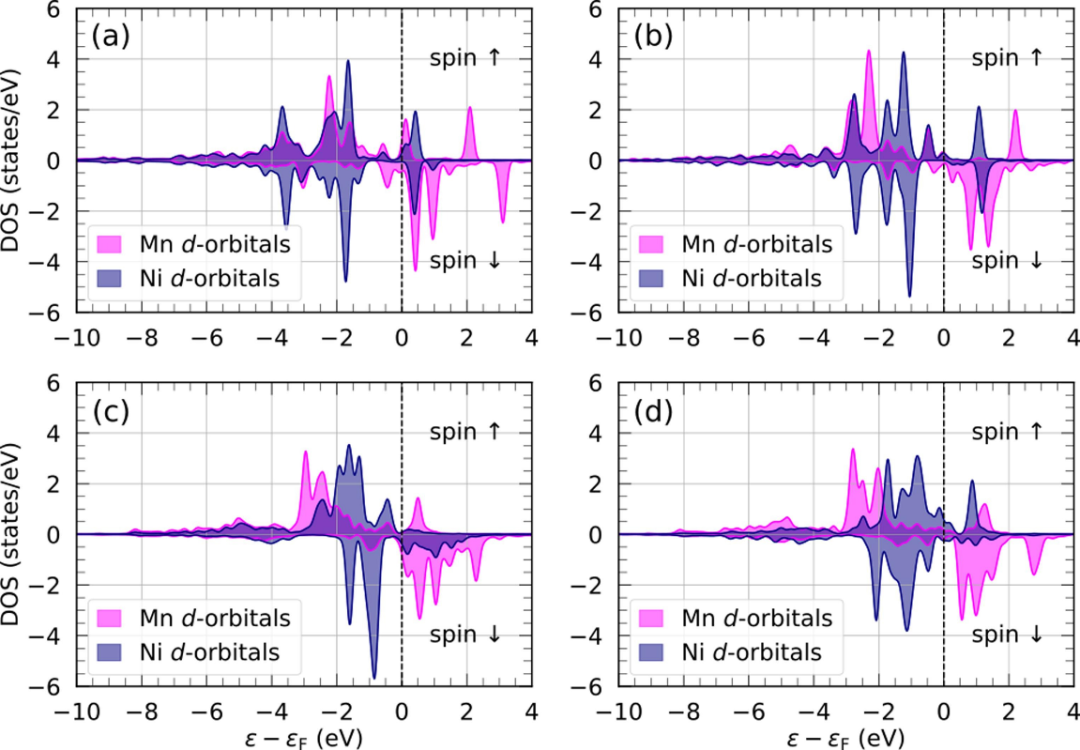
双原子协同催化:NiCu/rGO高效稳定析氧的机制与优势
下图展示了NiCu双原子催化剂(DACs)在还原氧化石墨烯(rGO)载体上的优化构型及其在氧析出反应(OER)中的催化机制,通过实验与理论计算的结合,揭示了双位点催化的多重优势。
这种双原子催化体系的核心价值在于其独特的协同效应,这种效应源于Ni和Cu原子之间的电子相互作用以及它们与rGO载体的紧密结合。
具体而言,Ni和Cu原子在rGO表面形成稳定的双原子结构,其结合能(-9.36 eV)显著高于单原子催化剂(Ni/rGO为-6.48 eV,Cu/rGO为-4.08 eV),这种强相互作用不仅提升了催化剂的稳定性,还通过电荷再分配优化了反应中间体的吸附能。
密度泛函理论(DFT)计算表明,Cu位点的OER过电位(0.33 V)远低于Ni位点(0.47 V)以及单原子催化剂(Ni/rGO为0.60 V,Cu/rGO为0.79 V),这一结果与实验数据高度吻合,证实了双位点催化在降低反应能垒方面的卓越性能。
双位点催化的优势进一步体现在其对反应动力学的优化上。OER是一个复杂的四电子过程,涉及多个中间步骤(*OH → *O →*OOH → O₂),而NiCu双原子催化剂能够通过协同作用分步优化这些步骤。
例如,Cu位点主要负责*OOH→O₂这一速率决定步骤,而Ni位点则可能辅助其他中间步骤的进行,从而整体上降低了反应的过电位。这种分工协作的机制得益于双原子之间的电子调谐,通过部分电荷转移(如从Cu到Ni)改变了活性位点的电子结构。
电荷密度差(CDD)分析显示,电子从金属原子向rGO载体转移,这种金属–载体相互作用不仅增强了催化剂的稳定性,还提高了电荷传输效率,这一点在电化学阻抗谱(EIS)中得到了验证——CuNi/rGO的电荷转移电阻显著低于单原子催化剂。
此外,双位点催化剂的电子结构特性对其催化活性具有决定性影响。通过分析分波态密度(PDOS),可以发现Ni和Cu的d轨道与rGO的p轨道在费米能级附近形成了强烈的杂化,这种杂化作用调节了金属原子的d带中心位置。
具体而言,Cu的d带中心在双原子催化剂中下移了1.52 eV,而Ni的d带中心也发生了类似但幅度较小的偏移(0.60 eV)。
d带中心的下移减弱了金属位点与反应中间体(如OH、O和*OOH)的吸附强度,从而避免了活性位点的过度阻塞,提升了催化反应的动力学性能。
这种电子结构的调控是单原子催化剂难以实现的,因为单原子位点缺乏相邻原子的协同作用,无法有效平衡中间体的吸附与脱附过程。