

说明:反应位点是化学反应优先发生的关键位置,其活性由电子结构(如Fukui函数、局部软度)和几何构型(间距、配位数等)决定。
通过DFT计算吸附能、活化能垒,结合NEB等过渡态搜索方法可定位位点。研究涵盖单/双原子催化剂、沸石酸位点等催化体系,及溶剂化效应、机器学习预测等方向,为催化剂设计与反应调控提供理论支撑。



什么是反应位点?
反应位点(Reaction Site)是理论计算中描述化学反应发生位置的核心概念,指分子或催化剂表面特定原子/基团因电子结构或几何构型特征,优先引发化学键断裂与形成的关键位置。其本质是能量最低反应路径上的核心节点,可通过量子化学计算或动力学精准定位。
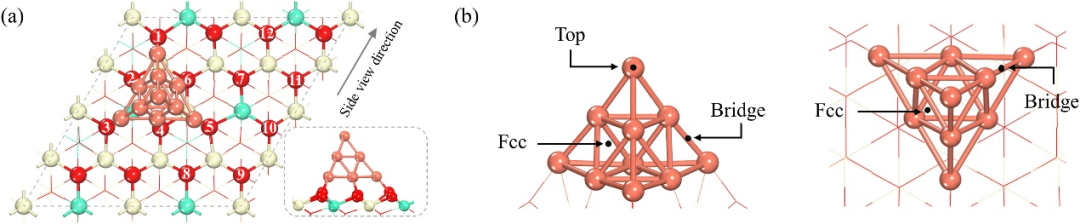
DOI:10.1016/j.jre.2024.09.015
反应坐标
反应坐标作为描述化学反应从反应物到产物演化过程的核心参数,本质上是在多维构型空间中定义的一条连续路径,其数值变化对应分子结构的逐步转变,而能量随反应坐标的变化曲线则直观呈现反应进程中的能量起伏。
在这一演化轨迹中,反应位点作为关键节点,特指过渡态结构或中间体的吸附位置,其本质是势能面上的驻点——过渡态对应能量曲线的最高点,是反应进程中最难跨越的能垒所在,决定了反应的速率限制步骤;中间体则对应局部能量极小值,是反应路径中相对稳定的中间状态,可能导致反应分支。
反应坐标与反应位点的关联体现在:反应位点的结构特征直接决定反应坐标的局部斜率与曲率,例如在亲核取代反应中,过渡态的五配位碳中心因键长的不对称分布,使得反应坐标在该区域呈现陡峭上升的能量趋势,反映出较高的活化能垒;而中间体的稳定化会使反应坐标在对应位置出现能量平台,为反应路径的调控提供可能。
在催化体系中,催化剂的活性位点通过与底物或中间体的特异性相互作用,可重塑反应坐标的能量分布——例如在铂催化的氢解反应中,金属表面的原子簇作为反应位点,通过吸附H₂形成活性氢物种,使反应坐标中H-H键断裂的过渡态能垒降低约0.3 eV,显著加速反应进程。
这种关联不仅是解析反应机理的基础,更为理性设计高效催化剂提供了依据——通过调控反应位点的电子结构或几何环境,可定向优化反应坐标的能量曲线,实现对反应速率与选择性的精准调控,从而深化对化学反应本质的理解并推动实际工业反应的效率提升。
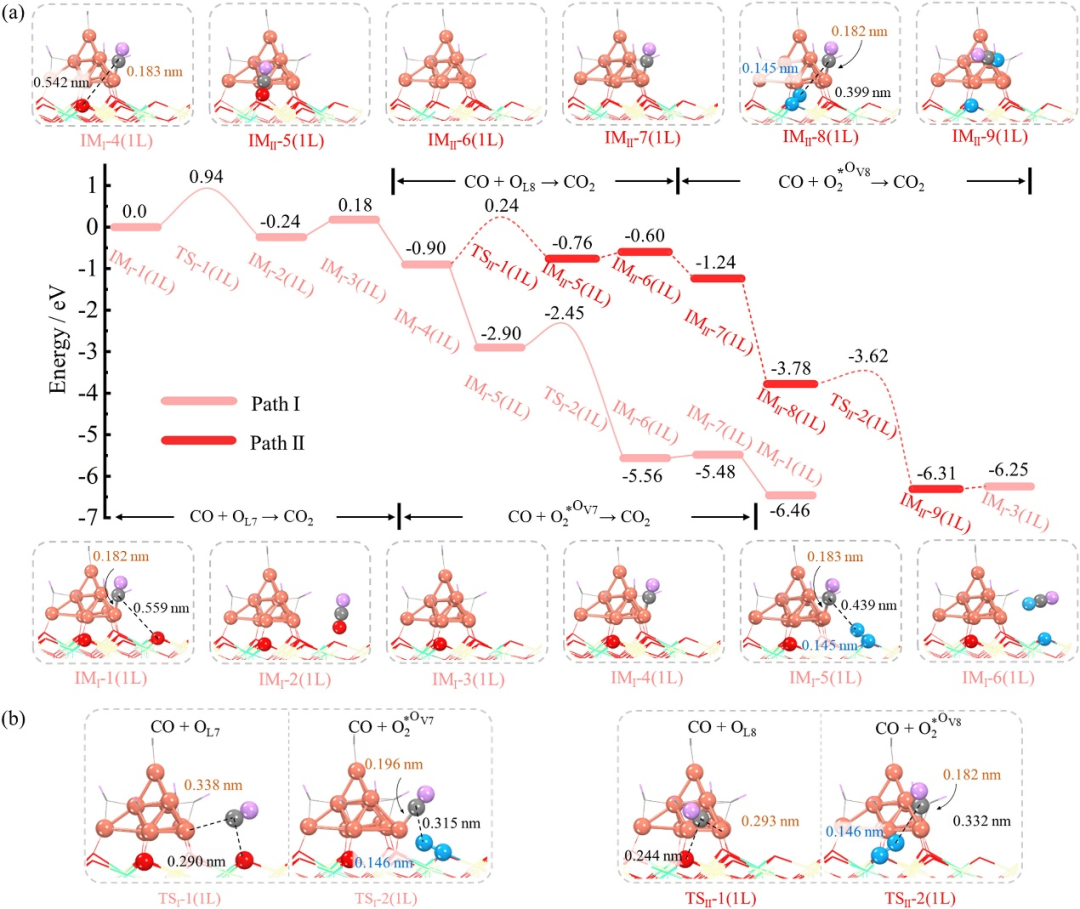
DOI:10.1016/j.jre.2024.09.015
电子结构特征
电子结构特征作为反应位点活性的核心决定因素,其影响通过亲电/亲核性与局部软度的量化表征得以明确。
亲电/亲核性可借助Fukui函数(f⁺/f⁻)或全局亲电指数进行定量描述:Fukui函数f⁺反映位点接受电子的能力,其值越高表明该位点电子云密度越低,越易被亲核试剂攻击,例如羰基化合物中C=O的碳原子因f⁺值显著高于周边原子,成为亲核加成反应的活性中心;而f⁻则表征位点给出电子的能力,电子云富集区域f⁻值较高,倾向与亲电试剂结合,如苯胺分子中氨基N原子的f⁻值达0.45 a.u.,使其在反应中表现出强定位效应。
局部软度作为另一个关键参数,描述电子云的极化难易程度,软度高的位点因电子易发生形变,在redox反应中更易参与电子转移——例如过渡金属催化剂中,低配位的Co²+位点较Co³+更易在OER反应中发生价态波动,其电子转移速率提升2–3倍。
几何构型约束对反应位点选择性的影响同样显著,催化剂表面金属位点的间距、配位数及分子的空间位阻通过限制反应路径实现选择性调控。
金属原子间距直接影响双分子反应的协同活化效率,如在CO₂加氢反应中,Cu催化剂表面Cu-Cu间距为0.25 nm时可同时吸附CO₂与H₂,而间距增至0.3 nm则因无法形成协同吸附导致活性下降40%;配位数通过改变金属d带中心调控中间体吸附能,例如Pt (111)表面高配位的terrace位点较低配位的step位点对*H的吸附能弱0.15 eV,使后者在HER反应中活性更高。
空间位阻的作用在择形催化中尤为突出,如ZSM-5沸石的十元环孔道仅允许直链烷烃进入并与内部酸位点作用,而支链烷烃因空间位阻无法扩散,使催化裂化产物中直链烃选择性达90%以上。
电子结构与几何构型的协同作用,共同决定了反应位点的活性阈值与选择性窗口,为通过原子级设计优化催化性能提供了从微观到宏观的理论依据。
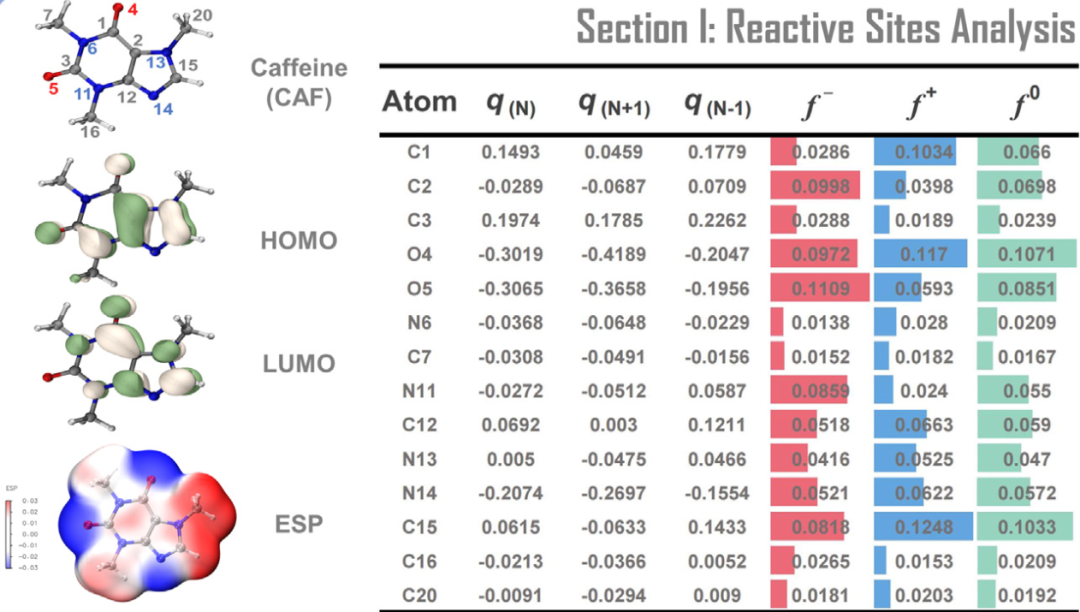
DOI:10.1016/j.trechm.2023.03.009
计算框架
反应位点的计算框架以密度泛函理论(DFT)为核心,通过量化吸附能与活化能垒实现对最优反应位点的精准定位——具体而言,通过比较催化剂表面不同位点对反应中间体的吸附能差异,可筛选出活性最高的位点。
例如在Pt基催化剂的氧还原反应中,计算表明step位点对*O的吸附能较terrace位点更接近火山图顶点,对应更高的催化活性;而活化能垒的计算则通过对比不同路径的能量,明确决速步对应的关键反应位点,如在CO₂电还原中,COOH形成的活化能垒在Cu-N₄位点低于Cu-N₃位点,证实N₄配位环境为更优反应位点。
过渡态搜索作为定位反应位点原子排列的关键步骤,主要依赖Nudged Elastic Band(NEB)或Intrinsic Reaction Coordinate(IRC)方法:NEB方法通过构建连接初态与末态的弹性带,在势能面上优化中间态结构以找到能量最高的过渡态,例如在甲烷解离反应中,NEB计算揭示过渡态的C-H键长较基态显著拉长,对应金属表面的三配位Fe原子为反应位点。
IRC方法则从过渡态出发沿能量梯度方向追踪反应路径,验证过渡态与初末态的连接性,确保反应位点的几何结构与实际反应进程一致。
溶剂化效应的引入需结合溶剂反应坐标,该坐标通过描述溶剂分子在反应中心两侧的配位数变化,量化溶剂对反应位点极性的调制作用——例如在酸性条件下,质子溶剂通过氢键网络使反应位点的局部介电常数提升20%,导致H吸附能降低0.15 eV,使计算结果更贴近实验测量值。
这些理论框架通过整合电子结构计算、动态路径分析与环境效应模拟,构建了从微观原子排列到宏观反应活性的完整关联,不仅能明确反应位点的具体原子组成与电子特征,还能揭示位点活性与结构参数的定量关系(如配位数每降低1,活化能垒降低0.08 eV),为通过原子级修饰(如掺杂、缺陷工程)优化反应位点性能提供了可操作的计算工具,推动催化机理研究从定性描述向定量预测的转变。
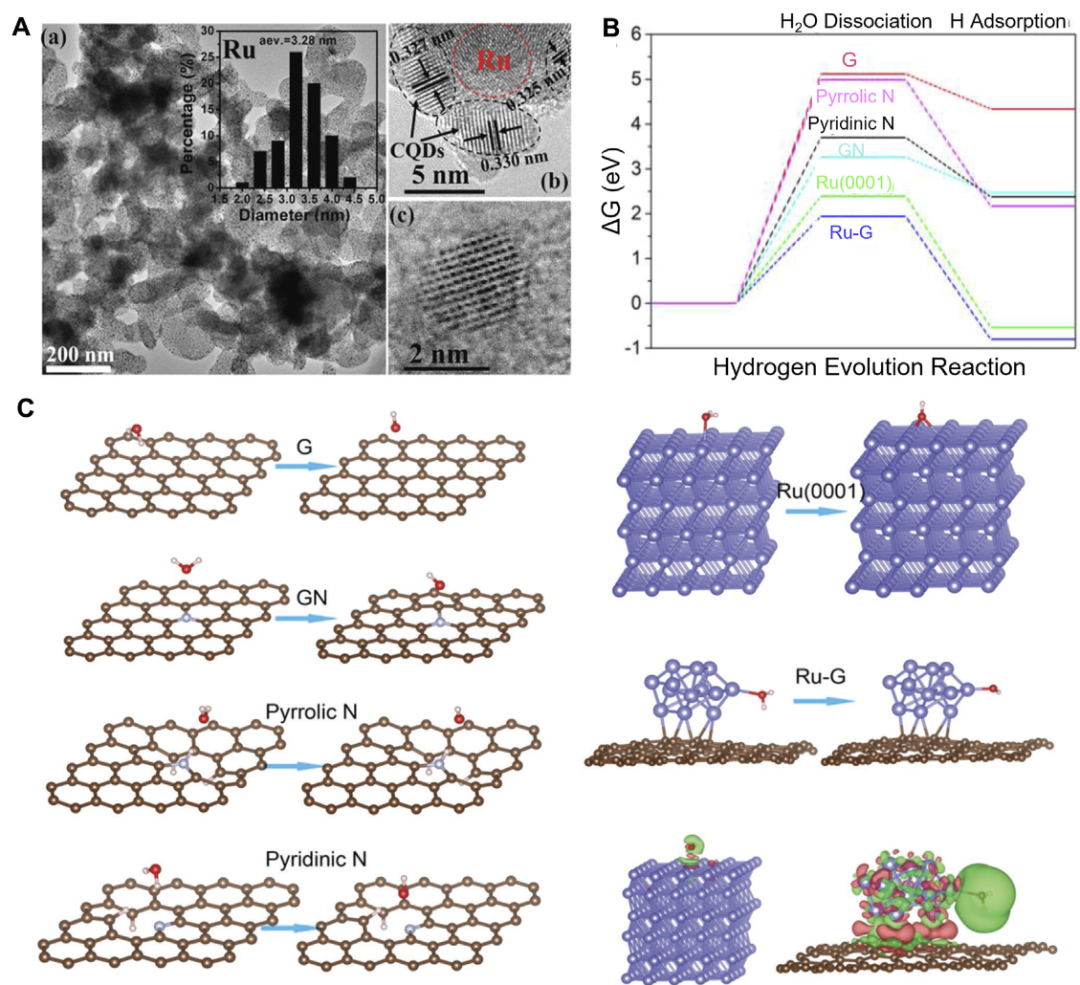
DOI:10.3389/fchem.2023.1286257
典型研究方向
反应位点的典型研究方向涵盖催化反应、溶剂化体系及机器学习辅助预测等领域,通过多维度方法解析位点活性与选择性的调控机制。
在催化反应研究中,单/双原子催化剂是重点,其通过两种金属原子的协同作用优化反应位点性能,研究中借助DFT计算金属位点对反应中间体的吸附能,预测C–C偶联等反应的活性,例如Fe/Co双位点体系中,Fe位点因电子结构特性对O₂的吸附能更低,主导O₂的活化过程,而Co位点则通过调节中间体的脱附能垒辅助产物生成,二者的空间分布与电子耦合显著提升催化效率。
沸石酸位点的研究则通过计算质子亲和能区分高活性的Brønsted酸位点与弱活性的硅醇巢,解释贝克曼重排反应中为何Brønsted酸位点能选择性催化肟转化为酰胺,其核心在于Brønsted酸位点的质子转移能力更强,与底物的相互作用能比硅醇巢高0.5 eV以上。
溶剂化体系中,溶剂反应坐标是描述位点环境影响的关键工具,“Pinching Coordinate”通过量化溶剂分子在反应中心两侧的配位数,刻画溶剂对反应位点的“挤压”效应,例如在Sₙ2反应中,溶剂分子的配位数变化会改变反应中心的电荷分布,使亲核试剂更易进攻反应位点。
“Plane-Inversion Coordinate”则通过追踪中心原子与特征平面的距离变化,描述空间位阻对反应位点可及性的影响,解释为何特定构型的分子更易在反应位点发生转化。
机器学习在反应位点预测中的应用日益广泛,其通过提取原子描述符训练模型,实现有机分子代谢位点的高效预测,例如针对CYP2C9酶底物的研究中,模型通过学习位点的电子云密度与空间结构特征,对代谢位点的预测准确率在前3位达到70%,大幅缩短了传统实验筛选的周期。
这些研究方向从不同角度揭示了反应位点的调控规律,为精准设计高效反应体系提供理论支撑。
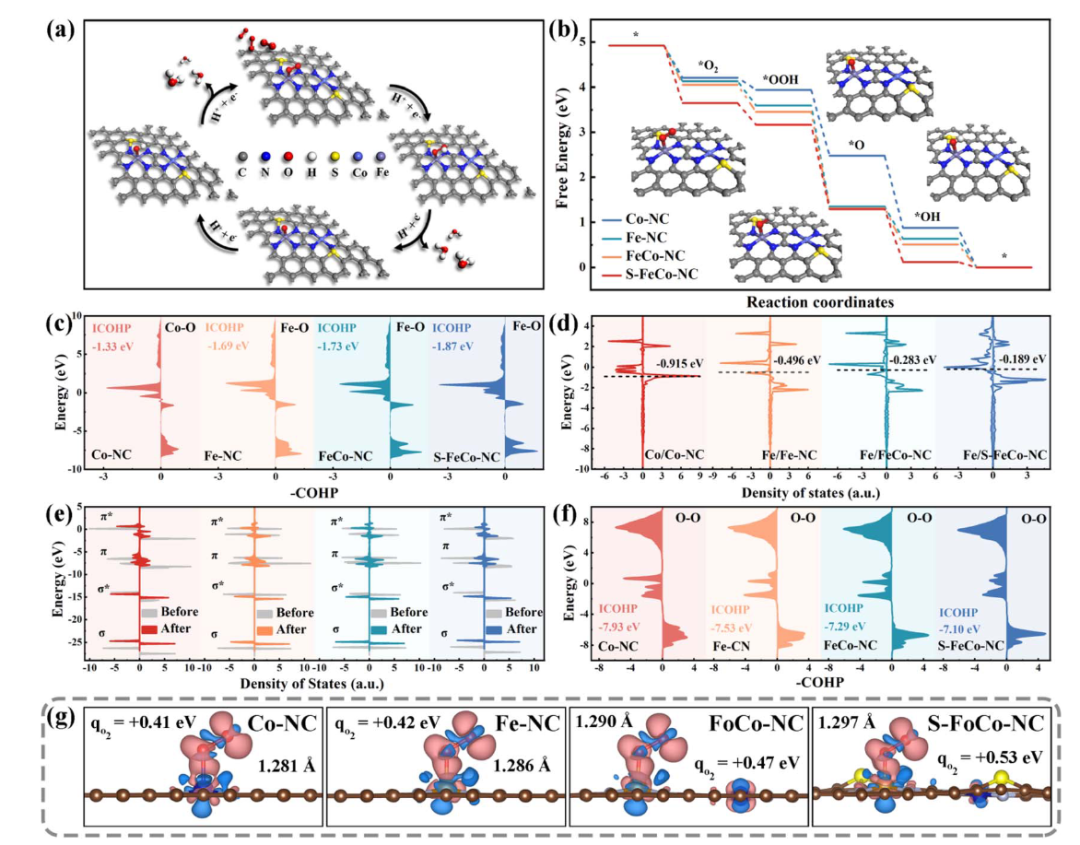
DOI:10.1039/d4sc03101f
单/双原子催化剂的应用
Zhang等以石墨炔(GDY)为载体,系统研究了过渡金属(TM=Sc、Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Zn)单原子(SACs)和双原子(DACs)催化剂的析氢反应(HER)活性,核心围绕反应位点的结构、稳定性、电子特性及催化机制展开。
反应位点的空间定位是其发挥作用的基础,GDY的18元环结构中,金属原子的最佳吸附位点为孔的角落(S₂),而非孔中心(S₁)或六元环中心(S₃),单原子催化剂在此通过TM-C配位形成稳定结构,双原子催化剂则在相邻S₂位点形成对称的TM-C配位,这种位点选择源于S₂处的几何约束与电子相互作用,可最大化TM与GDY的乙炔碳之间的键合强度,为HER提供固定的活性中心。
反应位点的稳定性是催化性能的前提,通过形成能和溶解电位评估显示,TM₂/GDY(如Ti、V、Fe、Co、Ni)的形成能更负,热力学稳定性更高,电子从TM向GDY的乙炔碳转移,3d/4s轨道贡献显著,这种电荷再分布增强了TM与载体的相互作用,使反应位点在电化学条件下不易分解,尤其Ni₂/GDY因均匀的TM-C键长和高对称性,位点稳定性最优。
反应位点的电子结构直接决定催化活性,费米能级附近的电子主要来自TM的3d轨道与GDY的C 2p轨道杂化,单原子催化剂中Cr、Mn、Fe、Co、Ni的氢吸附自由能在-0.295至0.178 eV,其中Ni₁/GDY的ΔG_H为-0.110 eV,接近理想值,因其位点的3d轨道与C 2p轨道杂化最强,实现氢吸附与解吸的平衡;双原子催化剂中,Cr-Ni位点虽ΔG_H整体为正,但电子密度分布更优,仍保持一定活性。
反应位点的 HER 机理进一步揭示其功能,Ni₁/GDY的Volmer-Tafel机理更占优,Tafel步骤能垒低于Heyrovsky步骤,因位点上吸附的H*通过相邻活性中心快速结合形成H₂,这与位点的几何对称性和电子流动性密切相关,确保氢在反应位点高效转化。
综上,GDY负载的TM单/双原子催化剂的反应位点,通过精准的空间定位、稳定的电子结构及优化的氢吸附特性,为高效HER提供了理想的活性中心,尤其Ni₁/GDY的位点性能最优,为低成本HER催化剂设计提供了理论依据。
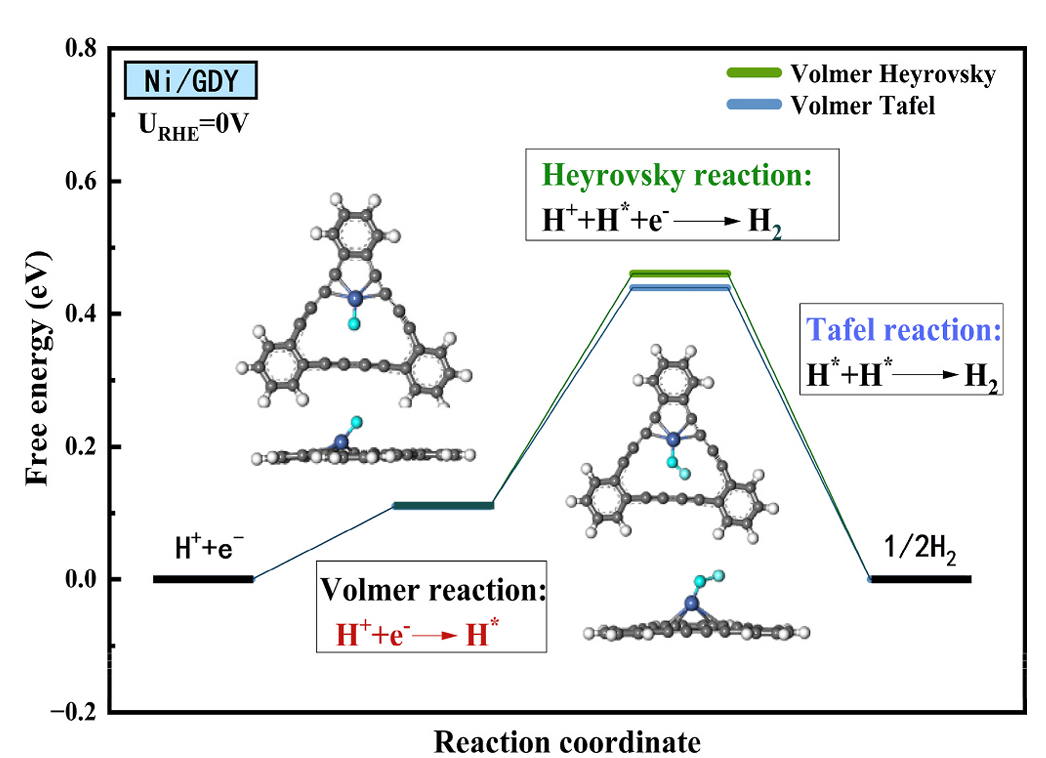
DOI:10.1016/j.ijhydene.2025.150144
总结
反应位点的理论研究正朝着动态识别、多尺度建模与人工智能辅助的方向发展,逐步构建起从原子尺度到宏观反应的完整认知体系。
动态位点识别需考虑原位条件下催化剂的结构弛豫,结合从头算分子动力学(AIMD)模拟,捕捉反应过程中表面重构对位点活性的影响,导致反应路径改变,AIMD通过追踪原子运动轨迹,可实时监测这种动态变化。
多尺度建模是连接微观与宏观的关键手段,QM/MM方法可处理酶活性中心等复杂体系,如漆酶的TNC位点,通过QM描述铜簇与底物的电子相互作用,MM处理周围蛋白质环境的空间约束,揭示质子迁移路径与位点活性的关联;微动力学模型则耦合DFT计算的能垒数据与反应动力学实验,通过验证活性位点的覆盖度与反应速率的关系,确认位点机制的可靠性。
人工智能辅助为复杂分子的反应位点预测提供了新工具,图神经网络(GNN)通过学习分子的拓扑结构与电子属性,可高效预测反应位点,如在有机合成中,GNN对药物分子反应位点的预测准确率已接近实验水平,大幅减少了筛选成本。
反应位点理论计算的核心在于关联电子结构、几何约束与反应路径,从原子尺度揭示化学选择性的物理本源。随着计算方法的迭代与硬件性能的提升,其在催化剂设计、药物代谢预测等领域的指导作用将日益凸显,推动从理论设计到实际应用的高效转化。