

密度泛函理论(DFT)是一种基于量子力学的计算方法,广泛应用于化学、材料科学、物理等领域,用于研究分子和材料的电子结构、化学键性质、反应动力学等。在化学键分析中,DFT提供了多种多维度的工具和方法,能够从轨道相互作用、电子密度分布、键能、键级、电荷转移等多个角度揭示原子间相互作用的本质。
DFT的基本原理与化学键分析的理论基础
DFT的核心是Kohn-Sham方程,通过构建一个非相互作用电子体系的虚拟轨道,计算真实体系的电子密度。在化学键分析中,DFT通过计算分子或材料的电子结构,揭示原子间的成键机制。例如,通过计算分子轨道的能级、电子占据情况、轨道重叠等,可以判断化学键的类型(如共价键、离子键、金属键)及其强度。
在DFT框架下,化学键的分析通常包括以下几个方面:
轨道相互作用分析:通过自然键轨道(NBO)分析,将分子轨道分解为定域化的自然键轨道,从而量化成键轨道和反键轨道的电子占据情况,进而计算键级(Bond Order)。例如,乙烷中的C-C单键键级为1,而乙烯中的C=C双键键级为2,这与实验测得的键能(346kJ/mol vs. 839kJ/mol)高度吻合。
电子密度拓扑分析:通过量子化学拓扑(QTAIM)和电子定域化函数(ELF)分析电子密度的分布,识别键临界点(BCP)和电子局域化区域。例如,在NaCl中,∇2ρ=+0.24a.u.表明其为离子键,而在苯分子中,ELF值η≈0.8表示其为π电子盆,具有芳香性。
键能与键长分析:通过计算分子的总能量和解离能,可以得到键能(Bond Energy),即断裂特定化学键所需的能量。例如,H2O中的O-H键能约为463kJ/mol,与实验值高度吻合。此外,键长(Bond Length)也是判断化学键强度的重要参数,通常与键级成反比。
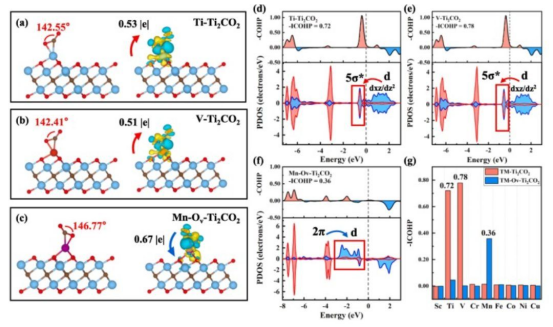
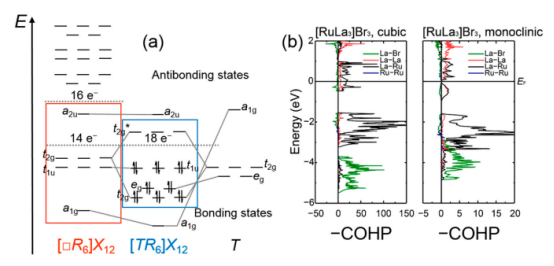
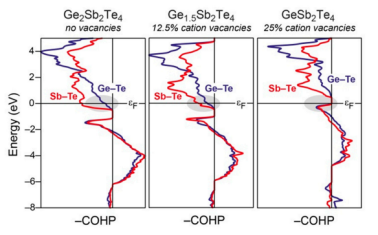
晶体轨道哈密顿布居(COHP)分析:COHP是一种专门用于周期性体系的化学键分析方法,通过计算原子间轨道相互作用的能量贡献,判断成键或反键作用。例如,在石墨烯中,C-C键的COHP值为正值,表明其为强共价键。
电荷与键级量化:通过Hirshfeld电荷、ADCH电荷等方法,可以量化原子间的电荷转移情况,从而判断化学键的极性。例如,在Li⁺-N3-体系中,Bader电荷分析表明Li⁺为正电荷,N3-为负电荷,符合离子键的特征。

DFT在不同类型化学键中的应用
共价键的分析
共价键是原子间通过共享电子形成的化学键,其强度和稳定性可以通过DFT的多种方法进行分析。例如,在苯分子中,通过ELF分析可以观察到π电子盆的形成,表明其具有芳香性。此外,通过NBO分析可以计算出苯环中C-C键的键级为1.5,与实验值一致。
离子键的分析
离子键是通过静电作用形成的化学键,其强度可以通过Bader电荷分析和电子密度分布来判断。例如,在NaCl中,Bader电荷分析显示Na⁺为+1.0,Cl⁻为-1.0,表明其为典型的离子键。此外,QTAIM分析显示NaCl的∇2ρ=+0.24a.u.,表明其为离子键。
金属键的分析
金属键是金属原子间通过自由电子云形成的化学键,其强度可以通过DFT的态密度(DOS)和COHP分析来判断。例如,在铜金属中,DFT计算显示其具有高度的电子离域性,COHP值为正值,表明其为强共价键。
氢键的分析
氢键是一种特殊的非共价相互作用,其强度可以通过DFT的电子密度分析和键能计算来判断。例如,在水分子中,O-H…O氢键的ρ=0.028a.u.,键能约为5kcal/mol,表明其为较弱的氢键。
DFT在实际研究中的应用案例
Ta4B18的化学键分析
在一项关于Ta4B18的研究中,研究人员通过DFT计算发现其具有完美的四面体结构,并通过AdNDP化学键分析发现其存在4个符合Hückel规则的6c-2e键,表明其具有球芳香性。此外,ELF分析显示Ta-B键的电子定域化函数值η达0.75,表明其为强共价键。
木质素的化学键分析
在木质素的选择性转化研究中,研究人员通过DFT计算解析了木质素的主要连接方式,并设计了高效的解聚体系。通过DFT计算,研究人员能够识别出木质素中的关键化学键,并预测其断裂路径。
1-羟基-2-萘甲酸的化学键分析
在1-羟基-2-萘甲酸(1H2NA)的研究中,研究人员通过DFT计算优化了其单体和二聚体的几何结构,并分析了其分子间氢键的形成。通过FT-IR和FT-Raman光谱与DFT计算结果的对比,验证了DFT在预测化学键性质方面的准确性。
DFT化学键分析的未来发展方向
随着计算能力的提升和算法的改进,DFT在化学键分析中的应用将更加广泛和深入。例如,机器学习与DFT的结合可以显著提高计算效率,使大规模体系的化学键分析成为可能。此外,多尺度模拟方法(如量子力学/分子力学混合方法)可以将DFT的原子尺度分析与宏观材料性能预测相结合,为材料设计提供更全面的理论支持。
总结
DFT作为一种强大的量子化学计算工具,能够从多个维度揭示化学键的本质。通过轨道相互作用分析、电子密度拓扑分析、键能与键长分析、COHP分析、电荷与键级量化等方法,DFT可以系统地解析共价键、离子键、金属键、氢键等不同类型化学键的形成机制和稳定性。在实际应用中,DFT不仅能够指导新材料的合成,还能为催化反应、药物设计、材料性能优化等领域提供重要的理论支持。随着计算技术的不断发展,DFT在化学键分析中的应用前景将更加广阔。