
VASP结构优化原理 准备工作 VASP输入参数 构建H2分子模型 总结
结构优化的核心目标是通过调整原子位置或晶格参数,使体系的总能量达到最小值。在VASP中,这一过程通常通过迭代算法实现,例如共轭梯度法(Conjugate Gradient)或准牛顿法(Quasi-Newton)。优化过程中,系统会根据当前的原子力和能量变化进行调整,直到满足预设的收敛条件(如能量变化小于设定值)。




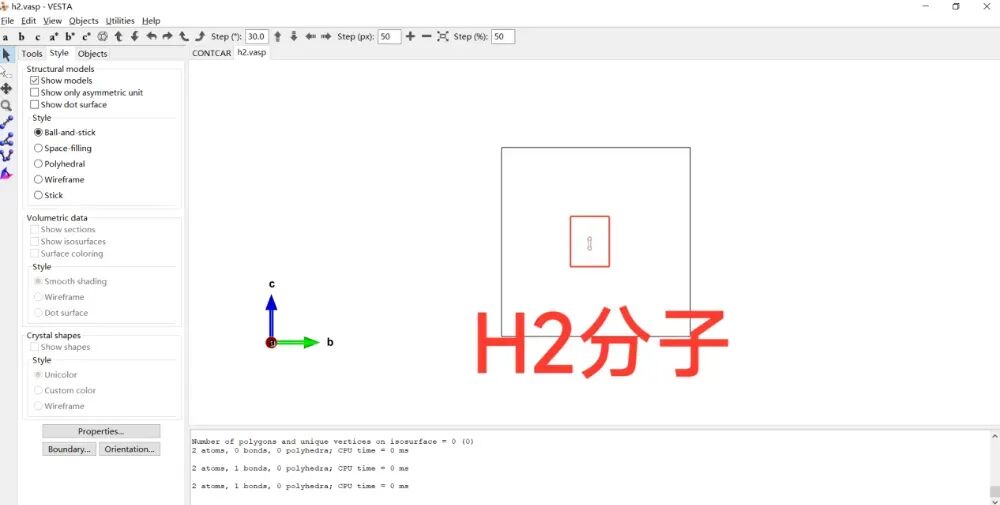
VASP教程 | VASP分子模型构建步骤!

声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!
赞 (0)
关于作者
VASP教程 | VASP如何构建分子模型?
上一篇
2025年11月3日 下午5:06
VASP | 理论计算常见问题解答-53
下一篇
2025年11月3日 下午5:45