

CeO₂的理论计算聚焦表面结构与缺陷、吸附及反应特性、电子调控等。通过DFT等方法解析氧空位形成能的晶面依赖性,量化吸附能与中间体稳定性,揭示电子结构调控机制;动力学模拟结合过渡态搜索与溶剂化效应,阐明反应路径。
研究热点包括形态–活性关系、双功能位点协同及高通量计算与机器学习应用,经典案例如Pd-CeO₂/OLC体系,为其在催化等领域的理性设计提供理论支撑。
CeO2能做什么计算
表面结构与缺陷形成能计算
二氧化铈(CeO₂)作为典型的稀土金属氧化物,其表面结构与缺陷化学的理论计算在催化、能源存储等领域具有重要意义,尤其是氧空位的形成机制与晶面依赖性研究,通过密度泛函理论为揭示活性位点本质提供了原子尺度的理论支撑。
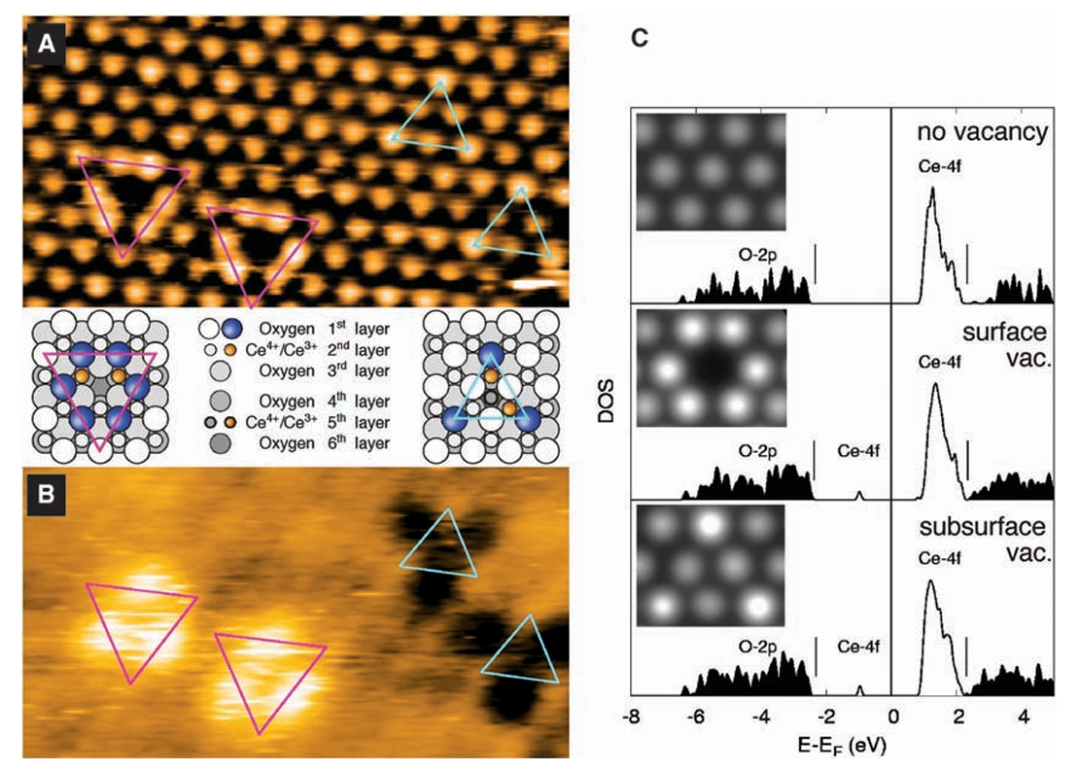
在氧空位形成机制的研究中,DFT计算表明,CeO₂表面与亚表面氧空位的稳定性差异源于Ce³⁺局域化电子态的调控作用:当移除晶格中的氧原子形成空位时,邻近的Ce⁴⁺离子捕获电子被还原为Ce³⁺,其4f轨道的电子局域化效应降低了空位形成能,使亚表面氧空位在热力学上更易生成。
以 (111) 晶面为例,亚表面氧空位的形成能显著低于表面空位,这是由于亚表面Ce原子的配位环境提供了更有利的电子再分配路径,使Ce³⁺的4f电子态在空位附近形成局域化能级,从而稳定缺陷结构。
这种电子局域化效应不仅影响空位的热力学稳定性,更使其成为催化反应的活性位点 —— 氧空位周围的Ce³⁺可通过可逆的价态变化(Ce³⁺↔Ce⁴⁺)促进氧物种的吸附与迁移,在CO氧化、水煤气变换等反应中发挥关键作用。
晶面依赖性研究通过DFT优化不同晶面(如 (111)、(110)、(100))的晶胞参数,量化了氧空位浓度与晶面结构的关系,发现氧空位稳定性序列为 (110) > (100) > (111)。
这一差异源于各晶面的原子配位环境与表面能差异:(110) 面的Ce原子具有五配位结构,移除氧原子后形成的配位不饱和Ce³⁺可通过相邻O原子的电子离域有效稳定,导致较低的形成能;而 (111) 面的六配位Ce原子在失氧后形成的配位缺陷需要更高的能量补偿,因此空位稳定性最差。
这些计算结果与实验观测到的晶面催化活性差异一致,例如 (110) 面因高氧空位浓度表现出更强的氧化还原能力,而 (111) 面的低空位浓度使其在需要稳定晶格氧的反应中更具优势。
CeO₂表面结构与缺陷形成能的理论计算,不仅揭示了氧空位形成的电子结构起源,更建立了 “晶面结构 – 空位浓度 – 催化活性” 的关联模型。通过DFT精确计算不同晶面的空位形成能与电子态分布,研究者可理性设计具有特定缺陷分布的CeO₂基催化剂,例如通过调控合成条件暴露高活性晶面(如 (110) 面)或引入掺杂剂降低目标晶面的空位形成能,从而优化其在氧还原、CO₂吸附等反应中的性能。
这种基于缺陷化学的理论研究,为 CeO₂在环境催化、燃料电池等领域的应用提供了从原子结构到宏观性能的全链条理论指导,推动了稀土金属氧化物催化剂的理性设计与高效开发。

DOI:10.1126/science.1111568
吸附能与反应中间体稳定性
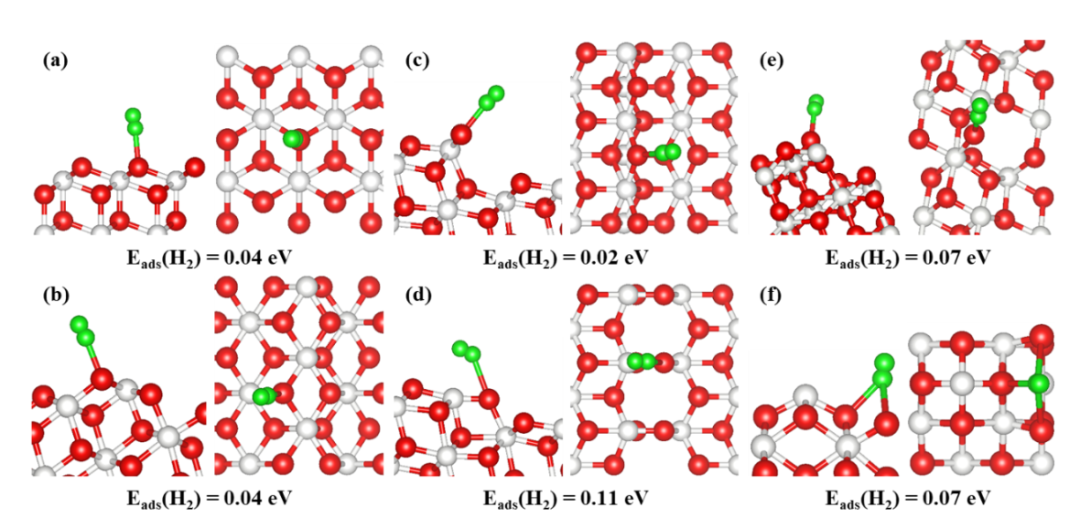
在CeO₂基催化剂的催化机制研究中,吸附能与反应中间体稳定性的理论计算为揭示表面反应动力学提供了关键依据。针对分子吸附构型,密度泛函理论(DFT)计算量化了H₂、CO、O₂等分子在CeO₂表面不同位点的吸附能。
结果显示吸附强度与晶面配位环境密切相关:例如H₂在CeO₂(111) 面的吸附能为0.04eV,而在高指数晶面 (223)-O-t上降至0.02eV,这种差异源于高指数晶面暴露的低配位 Ce 位点具有更强的电子给体能力,通过增强与分子的轨道杂化提升吸附作用,表明调控晶面取向可定向优化反应物的活化效率。
对于反应中间体的稳定性,通过吉布斯自由能计算进一步解析了电催化氧还原(ORR)与析氧(OER)反应的路径特征,这类计算揭示,不同中间体的ΔG差异直接决定反应的速率决定步骤,例如在CeO₂基催化剂上,若OOH中间体的ΔG显著高于其他步骤,则其形成过程为RDS,需通过掺杂或缺陷工程降低该步骤的能垒。
这些研究不仅建立了“吸附能–晶面结构”“自由能–反应路径” 的关联模型,更通过量化分析指导催化剂设计——如利用低配位Ce位点增强反应物吸附,或通过调控中间体ΔG优化决速步能垒,为提升CeO₂在能源转化反应中的催化活性提供了从原子吸附到反应动力学的全链条理论支撑。
DOI:10.1021/acscatal.1c04856
电子结构调控机制
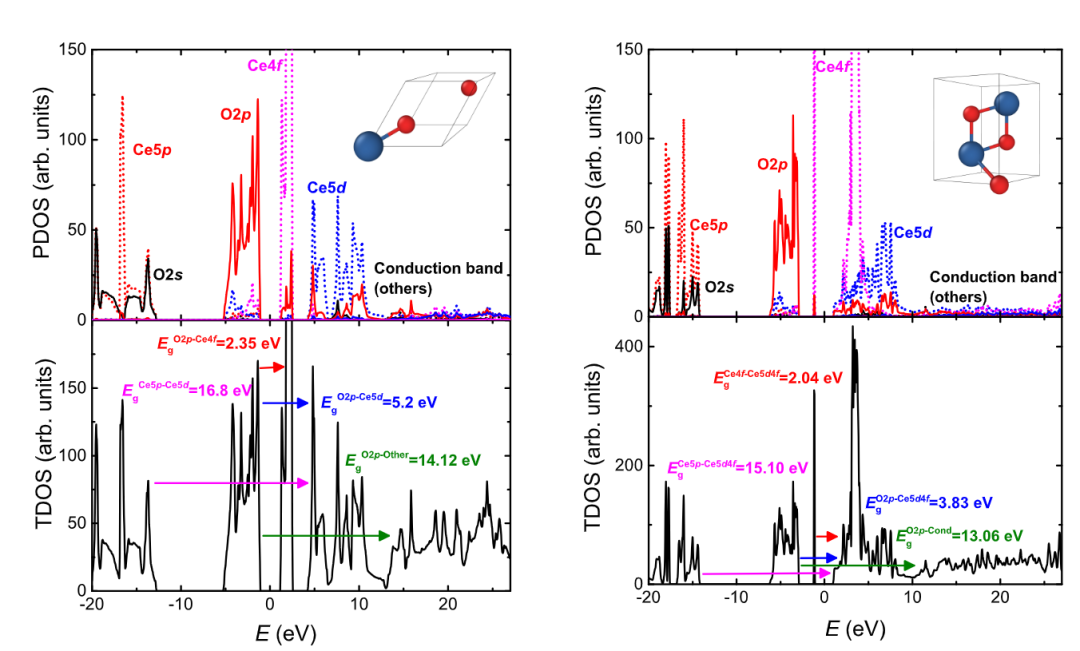
二氧化铈(CeO₂)的电子结构调控机制研究通过能带分析与电荷转移计算,揭示了其在催化体系中电子传输与活性位点优化的本质规律,为设计高性能催化剂提供了微观理论支撑。
在能带与态密度(DOS)分析中,投影态密度(PDOS)计算清晰展现了Ce-4f轨道与O-2p轨道的杂化特征:Ce⁴⁺的4f轨道在费米能级附近形成局域态,与 O²⁻的2p轨道杂化后,在导带底附近形成离域化的电子态,这种杂化程度直接影响材料的氧化还原性能。
以Pd-CeO₂/ 有序介孔碳(OLC)体系为例,CeO₂载体通过强金属–载体相互作用(SMSI)诱导Pd纳米颗粒的4d轨道电子离域,使复合体系的带隙从纯Pd的0.35eV 显著缩减至0.176eV,电导率提升一个数量级。这种带隙收缩源于CeO₂表面氧空位捕获Pd的d电子,形成Ce³⁺-Pdδ⁺界面态,增强了电子在金属–氧化物界面的传输效率。
电荷转移分析借助 Bader 电荷计算定量表征界面电子迁移行为,在Pd/CeO₂体系中,氧空位的存在促进了电子从Pd向CeO₂的定向转移(约0.23e⁻/Pd 原子),这种电荷再分配使Pd表面形成缺电子态(Pdδ⁺),而 CeO₂的 Ce³⁺浓度显著增加。
电子结构的这种变化有效优化了反应物的吸附行为:缺电子的Pd位点增强对CO等富电子分子的吸附,而Ce³⁺富集的CeO₂表面提升对O₂的活化能力。差分电荷密度图进一步显示,氧空位附近的电荷密度显著降低,形成电子 “活性区域”,促进Ce³⁺与吸附氧物种的氧化还原循环(Ce³⁺→Ce⁴⁺→Ce³⁺),这一过程在CO氧化反应中表现为决速步骤的能垒降低0.45eV。
CeO₂的电子结构调控本质上是通过轨道杂化与界面电荷转移重构材料的电子态分布,其核心机制包括:Ce-4f轨道的局域–离域平衡调控氧空位稳定性与氧化还原活性;金属–载体界面的电荷转移优化活性位点的电子构型,进而调节反应物吸附能与中间体稳定性。
这些理论计算不仅解释了实验中观察到的催化活性提升现象,更提供了普适性设计策略——通过引入异价金属掺杂(如Zr⁴⁺、Gd³⁺)或调控氧空位浓度,可精准调节Ce-4f轨道的电子占据数与杂化程度,实现对带隙宽度、电导率及表面吸附能的定向优化。
此类研究为CeO₂在汽车尾气净化、燃料电池等领域的应用提供了从电子结构到催化性能的跨尺度理论框架,推动了稀土金属氧化物催化剂的理性设计与高效开发。
DOI:10.1007/s10562-021-03650-4
反应路径动力学模拟
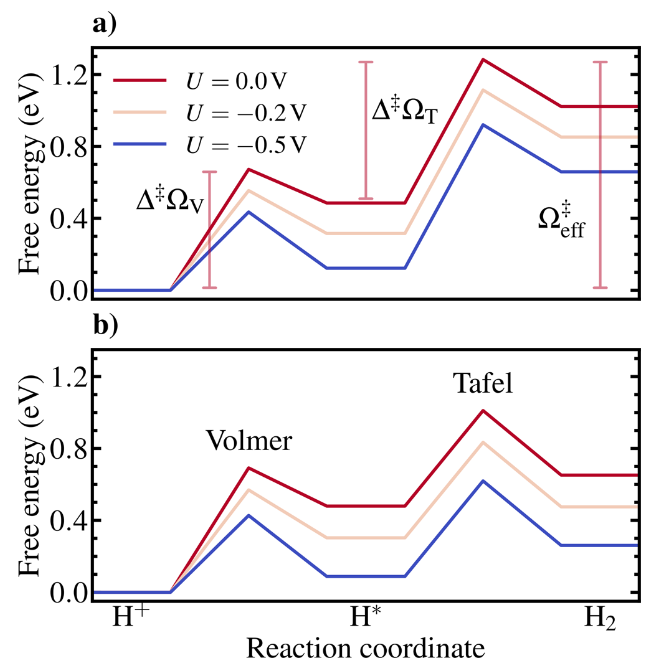
CeO₂基催化剂的反应路径动力学模拟通过精准刻画过渡态特征与界面环境影响,为揭示催化反应的微观机制提供了量化理论依据。
在过渡态搜索方面,采用nudged elastic band(NEB)方法可精确计算反应路径的能垒分布,以CO氧化反应为例,CeO₂(100) 晶面的过氧化物中间体(O₂²⁻)形成能垒仅为0.45eV,显著低于 (111) 晶面。
这种差异源于 (100) 晶面暴露的低配位Ce原子(五配位)与O₂²⁻的强相互作用——Ce-4f轨道与O₂²⁻的 π轨道杂化程度更高,稳定了带负电的过氧化物中间体,从而降低了过渡态能量。该结果解释了实验中 (100) 晶面比 (111) 晶面表现出更高CO氧化活性的现象,揭示了晶面配位环境对反应动力学的调控作用。
在溶剂化效应模拟中,通过显式溶剂模型或隐式溶剂修正,可有效模拟电化学界面环境对反应能垒的影响:例如在CeO₂催化的氧还原反应(ORR)中,水溶剂通过氢键与OH中间体作用,使该中间体的吸附能降低0.2eV,进而将决速步能垒从气相计算的0.8eV修正为0.6eV,更接近实验测得的动力学数据。
这种溶剂化修正的核心在于考虑了溶剂分子与反应中间体的非共价相互作用,以及溶剂对界面电荷分布的调制,使理论计算更贴近实际反应条件。
反应路径动力学模拟的价值不仅在于量化不同晶面、不同环境下的反应能垒差异,更通过揭示“过渡态结构–晶面配位–溶剂作用” 的关联,为优化CeO₂基催化剂性能提供了靶向策略——如优先暴露高活性 (100) 晶面以降低反应能垒,或通过调控溶剂化环境(如电解液pH)优化中间体稳定性,从而在CO氧化、ORR等反应中实现催化活性的定向提升,为多相催化体系的动力学优化提供了从理论模拟到实验设计的关键桥梁。
DOI:10.1021/acscatal.1c00538
核心计算方法
二氧化铈(CeO₂)的理论计算因Ce-4f电子的强关联性及氧空位缺陷的动态特性,需整合多种计算方法以精准刻画其电子结构与反应动力学。
密度泛函理论 + Hubbard U 修正(DFT+U)是处理Ce-4f电子强关联效应的核心方法,通过引入有效Hubbard参数,可准确描述Ce³⁺/Ce⁴⁺价态转变过程中的局域电子态,避免传统DFT对4f轨道离域化的错误描述,从而正确揭示氧空位形成时的电子再分配机制。
针对CeO₂带隙的精确计算,杂化泛函HSE06通过混合局域和非局域交换能,有效修正了广义梯度近似(PBE)对带隙的低估问题——实验测得CeO₂带隙约3.0eV,而PBE计算值仅为2.0eV,HSE06计算结果与实验值高度吻合,为准确分析光吸收、载流子激发等过程提供了可靠的电子结构数据。
从头算分子动力学(AIMD)模拟在研究CeO₂动态反应过程中发挥关键作用,尤其是Car-Parrinello MD方法,可在量子力学精度下追踪氧空位迁移的原子轨迹,揭示高温或电场条件下氧物种的扩散动力学。
例如,在质子耦合电子转移(PCET)反应中,AIMD能捕捉到 Ce³⁺/Ce⁴⁺价态变化与质子迁移的协同效应,量化缺陷迁移对催化活性的影响。
对于包含大量原子的大尺度体系,机器学习势函数结合神经网络力场,可显著加速缺陷扩散模拟,在保持较高精度的同时降低计算成本,为研究高浓度氧空位体系的热力学稳定性及扩散机制提供了高效工具。
值得注意的是,CeO₂的计算需严格考虑氧空位浓度、表面终端及自旋极化效应:氧空位浓度直接影响Ce³⁺的局域态分布,而表面终端决定活性位点的配位环境,自旋极化处理则避免因忽略Ce³⁺未成对电子导致的虚假电子态。
这些方法的协同应用,不仅解决了CeO₂电子结构描述的关键难题,更构建了从原子尺度缺陷形成到介观尺度动态过程的多尺度研究框架,为揭示CeO₂在CO氧化、氧还原等反应中的催化机理提供了坚实的理论支撑,推动其在能源催化与环境治理中的理性设计与应用优化。
DOI:10.1039/D1CP01810H
CeO2的研究热点
形态–活性关系
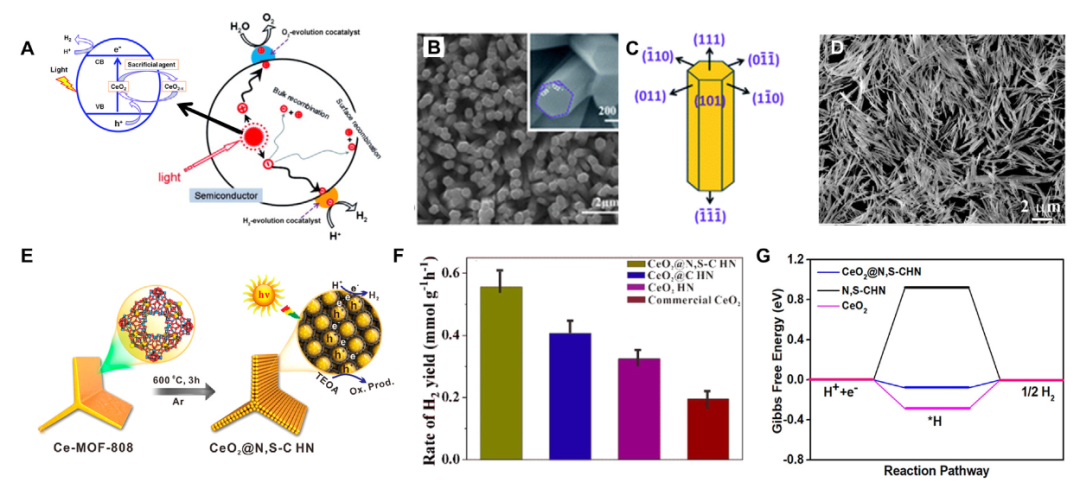
CeO₂的形态 – 活性关系研究通过晶面工程与异质结构设计,揭示了形貌调控对催化活性的关键影响。
棒状CeO₂因优先暴露 (110) 晶面,其表面氧空位浓度显著高于立方体 (100) 晶面,实验表明该结构的氧析出反应(OER)过电位降低85mV,这源于 (110) 面五配位Ce原子对氧空位的稳定作用,促进了活性氧物种的生成与迁移。
理论计算进一步证实,棒状结构的晶面配位环境使氧空位形成能降低,从而提升了表面氧化还原活性,这种形貌效应在CO₂加氢、HMF氧化等反应中也表现出优异的底物活化能力。
在二维异质结体系中,CeO₂/石墨烯复合结构通过界面电荷转移机制显著提升氧还原反应(ORR)性能。理论预测显示,石墨烯的高导电性与CeO₂氧空位的协同作用,使界面处形成Ce³⁺-Ov-C电荷转移通道,电子转移量可达0.3–0.5 e⁻,有效优化了 * OOH、*O 等中间体的吸附能,降低ORR决速步能垒0.25eV。
实验验证表明,该异质结在燃料电池中表现出优异的耐久性,50000次循环后仍保持69%的初始活性,远超传统Pt基催化剂,其性能提升与界面电子再分配诱导的中间体稳定性增强直接相关。
形态 – 活性关系的核心在于通过形貌调控实现活性位点的定向暴露与界面电子结构的优化:棒状CeO₂通过晶面工程最大化氧空位浓度,二维异质结则利用界面电荷转移构建高效催化路径。
这些研究不仅深化了对CeO₂催化机理的认知,更为设计高活性OER/ORR催化剂及能源转换材料提供了 “形貌–结构–性能” 的关联模型,推动其在燃料电池、金属空气电池等领域的实际应用。
DOI:10.3389/fchem.2022.1089708
双功能位点协同机制
CeO₂的双功能位点协同机制通过不同活性位点的电子结构互补,显著提升催化反应效率。
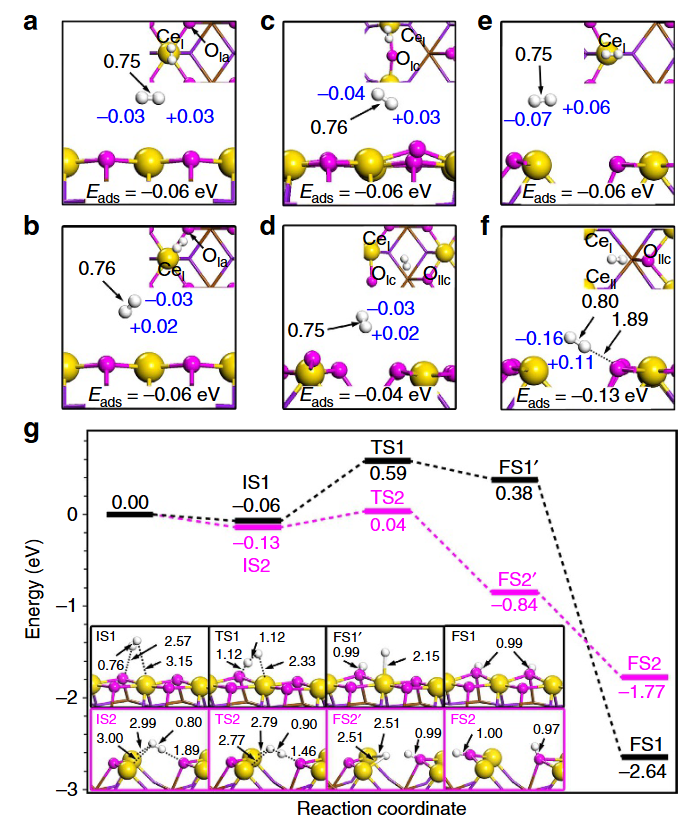
在固体受阻路易斯酸碱对(FLP)作用中,CeO₂(110) 表面的氧空位作为路易斯酸位点,与邻近的O²⁻路易斯碱位点形成空间匹配的酸碱对,二者协同作用可有效活化H₂分子——酸位点通过Ce³⁺的空轨道捕获H₂的σ电子,碱位点的O²⁻则通过孤对电子与H原子作用,使H₂解离能垒从单独位点的0.7eV降至0.32eV,大幅加速反应动力学。
而在金属–载体相互作用(MSI)中,Pt-CeO₂界面通过Pt的4d轨道与Ce的4f轨道杂化实现电子再分配,这种轨道耦合调节了Pt表面对H吸附中间体的结合强度,使氢吸附吉布斯自由能(ΔG_H)趋近于0eV,既避免过强吸附导致的脱附障碍,又防止吸附过弱影响解离效率,在氢氧化、加氢等反应中表现出优异的活性与选择性。
双功能位点协同的核心在于通过酸碱对的空间匹配与界面轨道杂化,实现对反应分子活化与中间体吸附的协同调控,为CeO₂基催化剂在能源转化反应中的高效应用提供了 “位点协同 – 电子调控” 的理论范式。
DOI:10.1038/ncomms15266
高通量计算与机器学习
在CeO₂基催化剂的理性设计中,高通量计算与机器学习的结合为高效筛选潜在体系提供了数据驱动的研究范式,其中Materials Project数据库的结构化信息成为关键支撑。该数据库整合了大量CeO₂基材料的晶体结构、能量数据及缺陷特性,为高通量计算提供了标准化的初始模型与参考基准。
研究中,通过高通量密度泛函理论(DFT)计算批量生成CeO₂基双金属催化剂的氧空位形成能数据,结合机器学习算法构建预测模型——以双金属掺杂浓度、原子占位、晶格畸变等为输入特征,氧空位形成能为输出目标,实现对未计算体系的快速预测。
这种方法显著提升了筛选效率:传统DFT单体系计算需数小时,而机器学习模型可在毫秒级完成预测,使研究范围从数十种扩展至数千种候选体系。筛选结果显示,Ni、Co等过渡金属掺杂可通过晶格应力调控Ce³⁺的稳定性,使CeO₂的氧空位形成能降低0.2-0.5eV,为实验合成高活性双金属CeO₂催化剂提供了明确靶点。
高通量计算与机器学习的协同,不仅突破了传统试错法的局限,更通过Materials Project数据库的跨研究数据整合,构建了 “结构–缺陷–活性” 的定量关联,推动CeO₂基催化剂设计从经验驱动向理论预测的范式转变。
DOI:10.3390/molecules26216485
经典案例:Pd-CeO₂/OLC
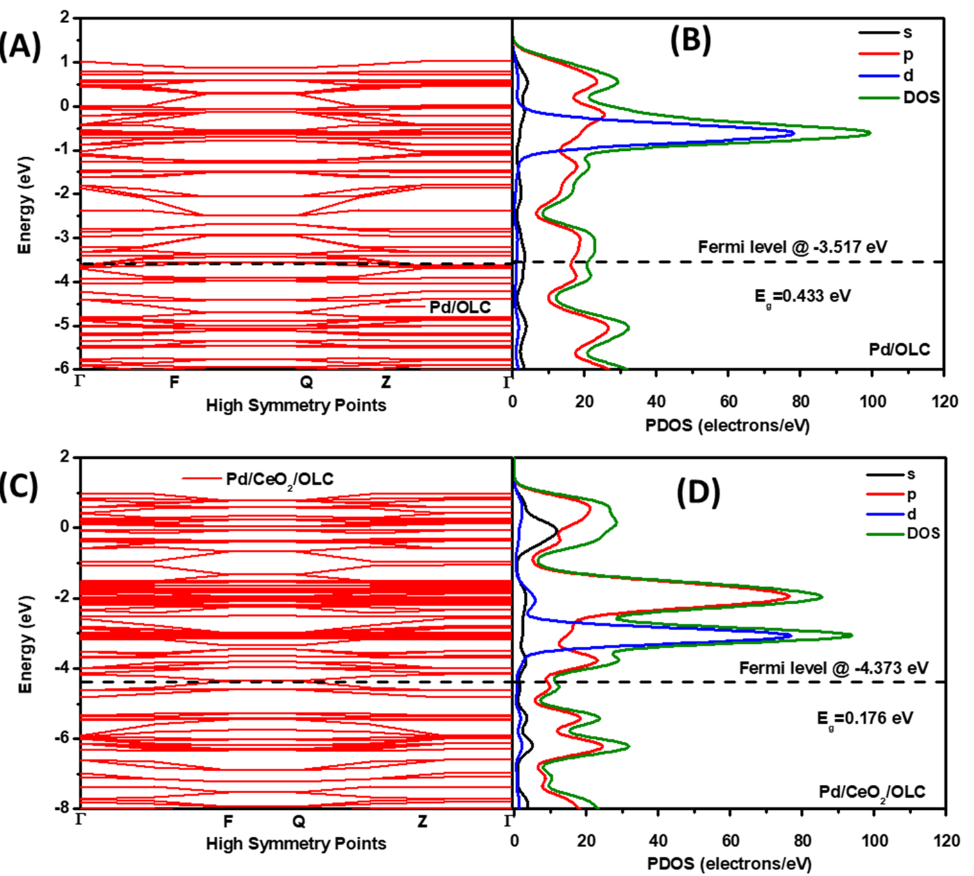
在Pd-CeO₂/ 洋葱状碳(OLC)催化剂用于氢氧化反应(HOR)的DFT研究中,研究者通过构建多层界面模型与电子结构分析,系统揭示了CeO₂载体对Pd催化活性的调控机制。
该研究以阐明CeO₂修饰OLC负载Pd体系的HOR活性起源为目标,构建了包含Pd (111)、CeO₂(111) 与OLC(002) 的三层界面模型,重点考察Pd-top(Pd位点顶部)、Ce-bridge(Ce原子桥位)及O-vacancy(氧空位)位点的分子吸附行为。
电子结构分析显示,CeO₂的引入显著改变了Pd的d轨道电子分布:投影态密度(PDOS)结果表明,Pd-4d轨道在费米能级附近的态密度显著增加,反映出CeO₂诱导的电子离域效应增强,这源于Pd与CeO₂界面的强相互作用——Ce-4f轨道与O-2p轨道在-2eV 能量处形成杂化峰,构建了高效的界面电荷转移通道。
Bader电荷分析进一步证实,Pd原子向CeO₂转移0.18个电子,而Ce原子获得0.12个电子,这种定向电荷转移使Pd表面呈现缺电子态,同时CeO₂中Ce³⁺浓度升高,为优化H*、OH * 等中间体的吸附能奠定了电子结构基础。
吸附能计算与反应路径模拟揭示了CeO₂对HOR动力学的关键优化作用:在 Pd/OLC体系中,H的吸附能为-0.51eV,而在Pd-CeO₂/OLC体系中降至-0.32 eV,吸附强度的弱化有利于H的脱附;OH的吸附能从-0.89eV优化至-0.67eV,表明CeO₂界面加速了碱性条件下 OH的形成与转化。
反应能垒计算显示,CeO₂的存在使H脱附能垒从0.75eV 显著降至0.48eV,这一变化源于氧空位附近的局域电子态对质子的稳定作用——氧空位作为活性位点,通过Ce³⁺的4f 轨道与H的s轨道杂化,降低了脱附过程的能量势垒。
从自由能图可见,决速步骤从Pd/OLC体系的H脱附转变为Pd-CeO₂/OLC 体系的OH形成,而后者的能垒更低,表明CeO₂通过界面电子调制重构了反应路径。
该研究的核心结论表明,CeO₂在Pd-CeO₂/OLC体系中发挥双重调控作用:一是通过界面电子转移使Pd表面缺电子化,优化H *吸附能至近理想值,平衡吸附强度以提升脱附效率;二是利用氧空位介导的质子转移,降低关键中间体的能垒,加速HOR速率。实验验证显示,该体系的HOR质量活性较纯Pd/OLC提升13倍,这与理论计算预测的能垒降低及吸附优化高度吻合。
从理论价值看,该研究构建了“金属–氧化物–碳载体” 界面的电子结构调控模型,揭示了CeO₂通过轨道杂化、电荷转移与缺陷协同效应提升催化活性的普适性机制。其方法论意义在于,结合多层界面模型、PDOS 分析与反应路径模拟,实现了从原子结构到宏观活性的跨尺度关联,为设计高效金属 – 氧化物复合催化剂提供了可复制的理论框架。
从应用前景看,该研究为碱性燃料电池阳极催化剂的优化提供了新方向,通过调控金属 – 载体界面电子结构与缺陷分布,有望突破传统Pt 基催化剂的活性与耐久性瓶颈,推动HOR催化剂从贵金属依赖向高效复合体系的转变。
总之,Pd-CeO₂/OLC体系的DFT研究不仅阐明了CeO₂在HOR中的具体作用机制,更展示了理论计算在揭示多组分催化体系协同效应中的独特优势——通过精准模拟界面电子行为与中间体动态过程,为高性能催化剂的理性设计提供了从微观机制到宏观性能的完整逻辑链条,推动催化研究从实验试错向理论指导的范式升级。
DOI:10.1021/acscatal.2c01863
总结
理论计算通过多维度方法创新实现深度发展:多尺度模拟融合密度泛函理论(DFT)与动力学蒙特卡洛(kMC),精准刻画实际电位下的表面重构过程;机器学习开发专用神经网络势函数,推动纳秒级从头算分子动力学(AIMD)模拟成为可能;动态界面模型构建电化学双电层,量化电位依赖的吸附能偏移规律。
当前研究已从静态结构优化迈向动态机制解析,为高性能电催化剂的理性设计提供原子尺度蓝图,未来需重点关注氧空位动力学、界面电荷转移及形态 – 电子耦合效应,以完善计算指导实验的闭环研究范式。