

MXene的理论计算涵盖材料本征性质、催化活性、反应路径及高通量筛选等方面。通过DFT分析电子结构、电荷分布及表面稳定性,借助吸附自由能、d带中心等揭示催化活性机制,结合自由能台阶图与过渡态搜索解析反应路径,高通量筛选加速高活性材料识别。
其在HER、ORR/OER、CO₂RR等反应中表现出潜力,为能源转化与碳循环领域提供理论支撑。
MXene能做哪些计算
材料本征性质
MXene的材料本征性质研究通过精准的电子结构分析与表面稳定性评估,构建了从原子尺度电子态到宏观环境稳定性的完整理论框架。
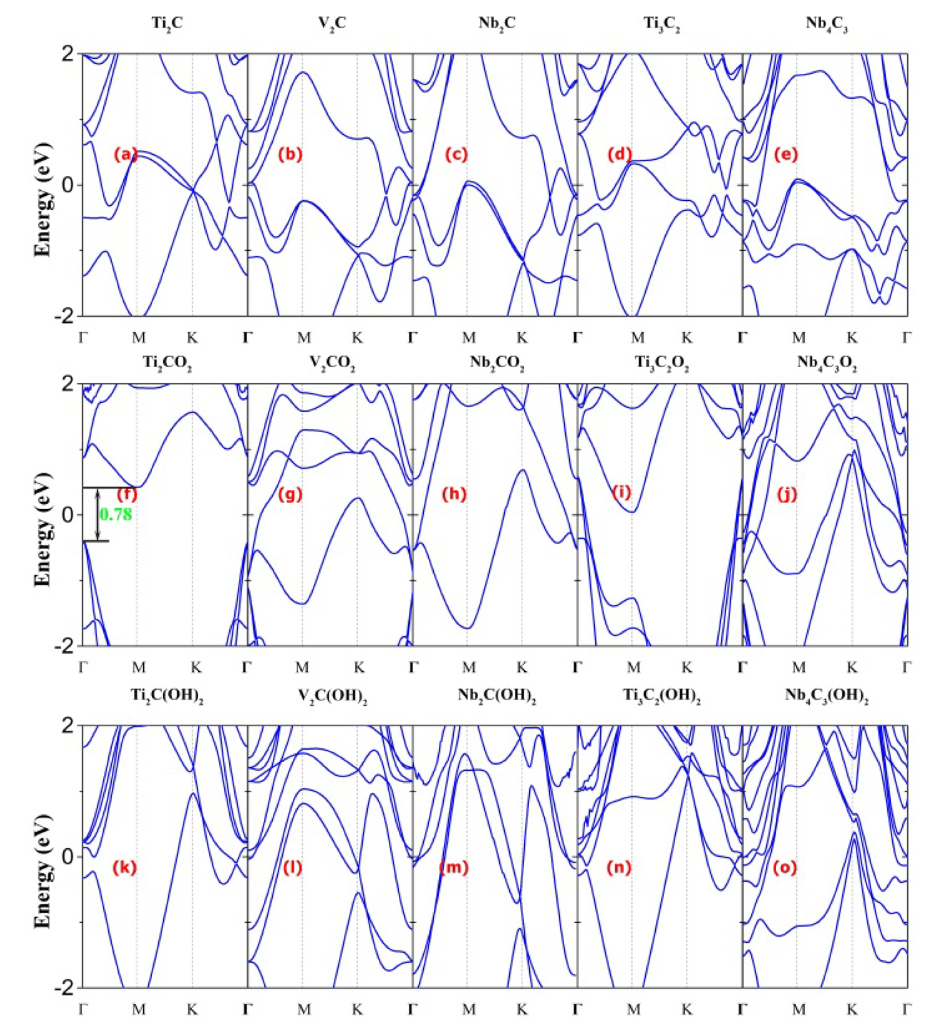
电子结构分析以密度泛函理论(DFT)为核心方法,通过能带结构与态密度(DOS)计算揭示其导电特性与电子态分布,例如TiCO的能带结构显示导带穿越费米面,呈现典型金属性,这一特征使其在电催化反应中具备高效电荷传输能力;态密度分析则可定位费米能级附近的主要贡献轨道,明确过渡金属d轨道与表面官能团p轨道的杂化程度,为理解电子转移机制提供依据。
电荷分布与转移研究借助 Bader 电荷分析量化表面官能团(–O/–OH/–F)的电荷再分配,电荷密度差分图显示,–O官能团因电负性较强,会从邻近金属原子吸引电子,形成局部电偶极矩,这种电荷分布直接影响表面对反应分子的吸附能与活化能力。
表面稳定性评估则通过Pourbaix图计算实现,基于不同pH值与电极电位下的吉布斯自由能变,确定MXene在特定反应条件下的稳定终止基团,例如TiC在氧析出反应(OER)的高电位、高pH环境中,–O终止的吉布斯自由能最低,成为热力学稳定相,这一结果与实验中观察到的表面重构现象一致。
Pourbaix图的构建需整合DFT计算的形成能与溶剂化效应校正,通过量化环境因素的影响,为预测MXene在复杂电解液中的稳定性提供热力学依据。
这些计算不仅揭示了MXene本征的电子特性与表面化学状态,更建立了 “电子结构–表面官能团–环境稳定性” 的关联模型,为选择合适的MXene体系用于特定催化反应提供了理论指导,避免了实验中的盲目筛选。

DOI:10.1021/acscatal.6b02754
催化活性
MXene的催化活性研究通过吸附自由能计算与电子结构调控机制分析,揭示了其在能源转化反应中的活性起源与优化路径。
吸附自由能是评估催化活性的核心热力学描述符,在析氢反应(HER)中,ΔG_H*(氢吸附自由能)的计算需整合DFT吸附能、零点振动能(ZPE)校正与熵变项,理想催化剂的ΔG_H应接近0eV,MXene中部分体系的ΔG_H可达-0.05eV,接近这一理想值,表明其优异的HER潜力。
在氧还原(ORR)与氧析出(OER)反应中,通过计算O、OH、OOH等中间体的吸附能,利用标度关系构建活性火山图,可量化不同MXene的催化活性,例如TiCO的OH吸附能适中,在火山图中处于高活性区域。
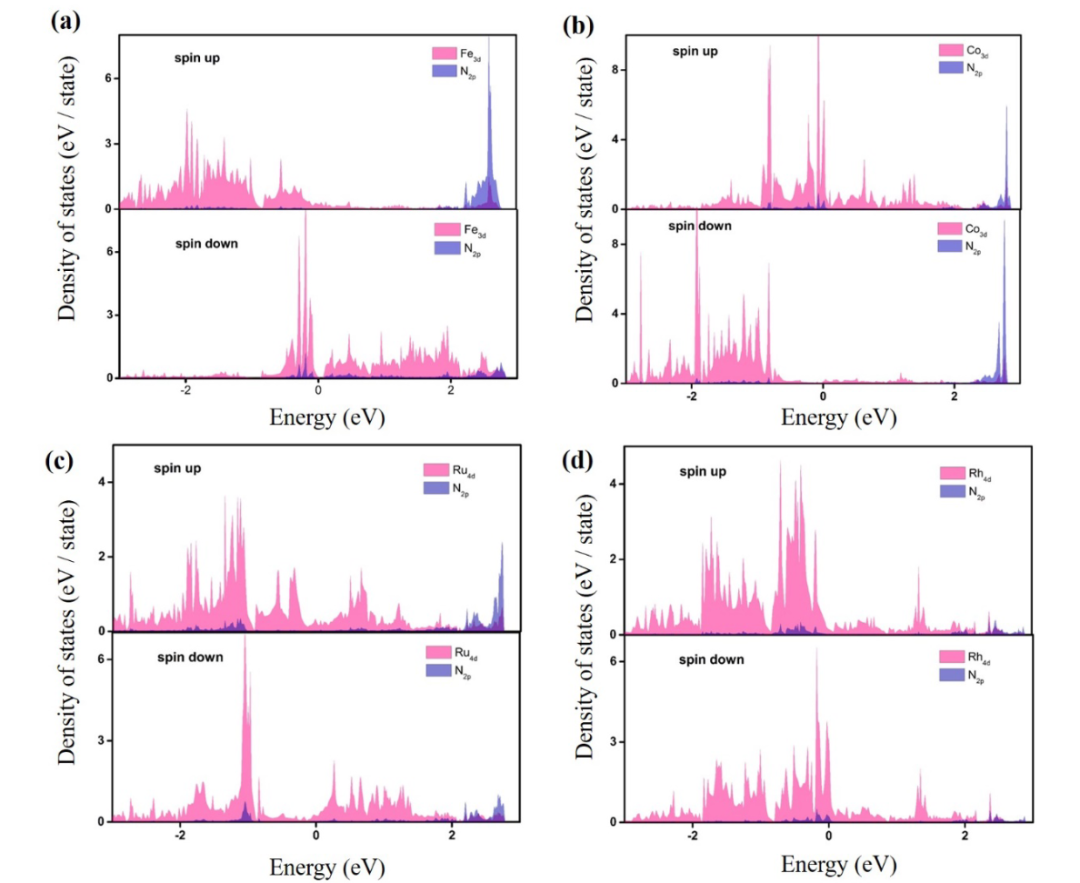
d带中心理论进一步揭示了活性调控的电子机制,过渡金属的d带中心位置与反应分子的吸附强度直接相关,d带中心上移会增强金属与吸附质的轨道杂化,提升吸附能,如Fe单原子修饰的TiCO体系中,Fe的3d轨道与Ti的3d轨道发生杂化,使d带中心向费米能级移动,显著增强对N₂分子的吸附与活化,为氮还原反应(NRR)提供高活性位点。
这种电子结构调控不仅适用于单原子修饰,掺杂、缺陷工程等手段也可通过改变d带中心实现活性优化,例如W掺杂Mo₂CTx使d带中心上移0.2eV,增强了对H的吸附能力,ΔG_H * 从-0.15eV优化至-0.08eV。
吸附自由能与 d 带中心的关联研究,将宏观催化活性追溯至微观电子态,使 MXene 的活性优化从经验尝试转变为靶向设计,例如通过调控表面官能团类型(–O/–OH)改变d带中心位置,或通过合金化调整过渡金属组成,实现对吸附能的精准调控,为开发高性能MXene基催化剂提供了明确的理论依据。
DOI:10.1016/S1872-2067(18)63197-3
反应路径
MXene催化反应路径的理论模拟通过吉布斯自由能台阶图与过渡态搜索,揭示了从反应物到产物的完整动力学过程与决速步骤。
吉布斯自由能台阶图通过量化多步反应中各中间体的自由能变化,构建反应能量景观,例如在析氢反应(HER)中,Volmer-Heyrovsky路径涉及H⁺吸附与电化学脱附两步,自由能台阶图可明确两步的能垒差异,确定决速步;而Tafel路径的自由能变化则需考虑双原子吸附的协同作用。
在氮还原反应(NRR)中,N₂逐步质子化生成NH₃的过程通过自由能台阶图显示,NNH形成步骤的自由能垒最高,成为决速步,这一结果与实验中观察到的高过电位现象一致。
过渡态搜索采用爬坡弹性带法(CI-NEB),通过构建初始态与终态间的中间映像,优化得到鞍点结构与能垒高度,例如在CO₂还原反应中,COOH→CO的过渡态能垒计算显示,MoCO-O1体系的能垒为0.5eV,而MoCO-O2因COOH吸附过强,能垒升至0.8eV,解释了两者催化活性的差异。
过渡态的几何结构分析可揭示反应的微观机制,如 COOH→CO过渡态中,C-O键长从1.26Å增至1.32Å,表明键的活化与断裂过程,而金属位点与C原子的距离缩短则反映了催化中心的电子转移作用。
这些计算不仅明确了各基元反应的能垒分布,更通过对比不同 MXene 体系的路径能垒,筛选出具有低决速步能垒的高效催化剂,例如Ti₃C₂O₂在ORR中OOH→O的过渡态能垒比Ti₃C₂F₂低0.3eV,表现出更优活性。
反应路径的模拟为理解MXene的催化选择性提供了关键依据,如在NRR与HER的竞争反应中,通过比较两者的决速步能垒,发现MXene在-0.5V(vs RHE)时NRR能垒更低,可抑制HER,提升NH₃产率,为实验条件优化提供了理论指导。
DOI:10.1007/s12209-020-00235-x
高通量筛选
MXene的高通量筛选通过构建结构化数据库与描述符关联,实现了从海量候选体系中快速识别高活性材料的目标。
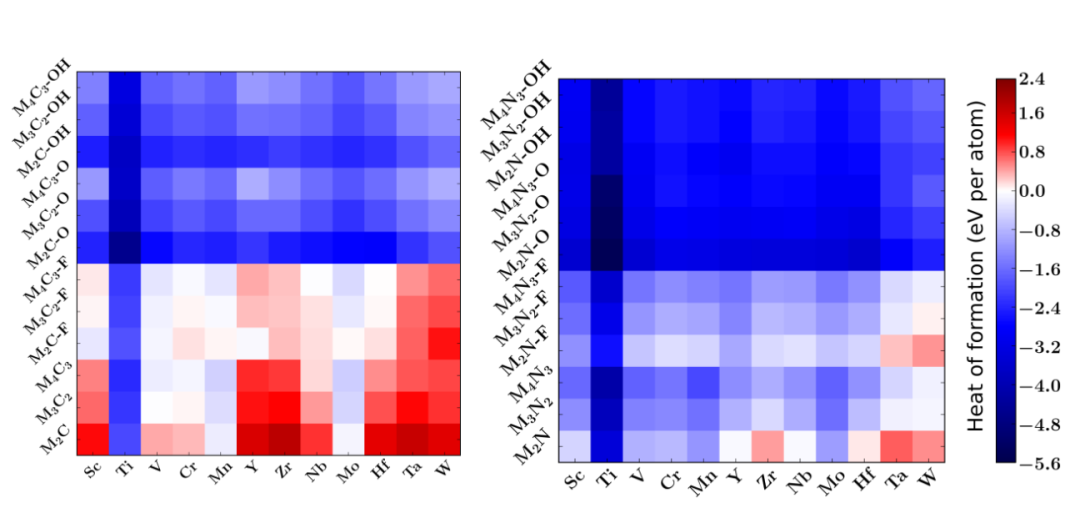
数据库构建涵盖72种MXene,系统计算其氢吸附自由能(ΔG_H*)与层数、终止基团(–O/–F/–OH)的关联性,例如单层MXene因表面原子占比更高,ΔG_H普遍优于多层体系。
–O终止MXene由于与H的吸附强度适中,ΔG_H多分布在火山图顶点附近,而–F终止因电负性过强导致吸附过弱,ΔG_H正值较大。数据库的建立需整合高通量DFT计算与自动化结构优化流程,通过标准化的计算参数确保数据可比性,为后续机器学习建模奠定基础。
描述符关联则通过统计分析发现ΔG_H与表面功函数的线性关系,表面功函数反映材料表面电子逸出能力,与表面电荷密度直接相关,这种线性关系表明可通过测量或计算表面功函数快速预测ΔG_H*,无需复杂的吸附能计算,大幅提升筛选效率。类似地,OER活性与d带中心、ORR过电位与表面氧空位形成能等描述符关联也被相继发现,形成了多维度的快速评估指标。
高通量筛选的价值不仅在于高效识别活性体系,更在于揭示潜在的构效关系,例如发现层数每增加1层,–O终止MXene的ΔG_H平均升高0.03eV,这一规律指导实验通过控制剥离程度调控层数以优化活性。
此外,结合机器学习算法,可基于数据库训练预测模型,输入MXene的组成、结构参数即可输出催化活性,例如对1000种未计算的MXene衍生物,模型预测ΔG_H的误差可控制在0.05eV以内,为新型MXene的设计提供了数据驱动的理论工具,加速了从基础研究到应用开发的转化进程。
DOI:10.1021/acs.jpcc.7b05270
MXene典型计算内容
析氢反应(HER)
MXene在析氢反应(HER)中的理论计算通过系统化的流程设计,揭示了其活性规律与优化策略。
计算流程首先聚焦H吸附位点的优化,基于MXene的晶体结构特征,评估顶位、桥位、空心位等可能的吸附构型,通过结构弛豫计算各位点的吸附能,确定最稳定吸附模式,例如MoCT中H更倾向吸附于Mo原子顶位,而Ti₃C₂O₂中空心位为最优位点。
随后计算氢吸附自由能(ΔG_H*),以贴近实际反应条件。最后通过绘制ΔG_H火山图,将不同MXene的活性定量化,火山图顶点对应ΔG_H≈0 eV,此时H的吸附与脱附达到平衡,催化活性最高。
关键计算结论揭示了表面官能团与过渡金属种类对HER活性的决定性影响:–O终止MXene的ΔG_H普遍优于–OH/–F终止,原因在于–O的电负性适中,与H的相互作用强度接近理想值,而–F因电负性过强导致H吸附过弱,–OH则因氢键作用使吸附过强。
过渡金属为Mo的MXene表现出优异活性,其ΔG_H = -0.05eV,与Pt的-0.09 eV非常接近,这源于Mo的d带中心位置适中,与H的1s轨道杂化程度理想,促进电子转移与H-H键形成。
此外,层数效应研究显示单层MXene的ΔG_H *比四层体系低0.08eV,因单层结构暴露更多表面原子,且量子尺寸效应使电子态密度在费米能级附近增强,提升了催化活性。
这些计算结果不仅明确了MXene在HER中的活性起源,更提供了靶向优化路径,例如通过表面处理引入–O官能团、选择Mo基MXene或制备单层结构,可显著提升HER性能,为实验合成高活性MXene基HER催化剂提供了清晰的理论指导,实验中基于这些结论制备的MoCT催化剂在酸性电解液中表现出10mA/cm²电流密度下的过电位仅为80mV,与理论预测高度吻合。
DOI:10.1002/aenm.202103867
氧还原 / 析出反应(ORR/OER)
MXene在氧还原(ORR)与氧析出(OER)反应中的理论计算通过活性描述符构建与趋势分析,揭示了其在电催化能量转换中的应用潜力。
ORR/OER的核心描述符为过电位(η),其中ORR过电位(η_ORR)的计算基于中间体自由能差,OER过电位(η_OER)则通过类似方法计算,反映反应动力学的难易程度。
计算过程需先优化各中间体在MXene表面的吸附构型,例如OH倾向通过O原子与MXene表面金属原子成键,OOH则可能形成双齿吸附模式,再通过DFT计算结合溶剂化效应与电位校正,得到各中间体的自由能,进而确定决速步与过电位。
活性趋势分析显示,单原子修饰可显著优化MXene的ORR/OER性能,如TiCO@Fe单原子体系中,Fe的3d轨道与O中间体的2p轨道发生强杂化,降低了OOH→O步骤的自由能差,使η_ORR从0.52V降至0.35V,接近商业Pt/C催化剂的活性。
表面官能团对活性的影响同样显著,–O终止的MXene在OER中表现更优,因–O可通过质子耦合电子转移稳定O与 * OH中间体,而–F终止则因强电负性抑制中间体吸附,导致过电位升高。
过渡金属种类的影响体现为,具有中等d带中心的金属(如Co、Ni)掺杂MXene,其d带中心与O的2p轨道能级匹配度更高,利于电子转移与键活化,使η_OER降低0.1-0.2V。
这些计算不仅量化了MXene的ORR/OER活性,更建立了 “表面结构–电子态–过电位” 的关联模型,为设计高活性催化剂提供了明确方向,例如通过单原子修饰调控d带中心,或选择–O终止的Co掺杂MXene体系,可在金属–空气电池、电解水等器件中实现高效能量转换。
DOI:10.1002/sstr.202200354
CO₂还原反应(CO₂RR)
MXene在CO₂还原反应(CO₂RR)中的理论计算通过反应路径分析,揭示了其对CO₂转化的活性与选择性机制。
计算聚焦CO₂转化为CO、CH₄、HCOOH等产物的可能路径,通过优化各中间体的吸附构型,构建吉布斯自由能台阶图,量化各步反应的能垒,例如MoCO-O1体系的路径分析显示,其中COOH→CO步骤的自由能垒为0.75eV,成为决速步,这一结果与实验中观察到的以CO为主要产物的现象一致。
过渡态搜索进一步明确了决速步的微观机制,COOH→CO过渡态中,C-O键长从1.26Å增至1.32Å,表明键的活化过程,而Mo原子与C原子的距离缩短0.05Å,反映金属中心向C原子的电子转移,这种电子转移增强了C的亲核性,促进O原子的离去。
不同MXene体系的活性差异源于对COOH中间体的吸附强度,MoCO-O2因表面O官能团密度高,与COOH形成强氢键作用,导致COOH吸附能过负(-1.8 eV),使COOH→*CO的能垒升至1.1eV,活性显著低于MoCO-O1;而Ti₃C₂(OH)₂通过适度的Ti-O键相互作用,COOH吸附能为-0.9eV,能垒降至0.5eV,表现出更优活性。
选择性调控机制研究显示,MXene的表面电荷分布可影响CO₂RR与HER的竞争,例如N掺杂MXene表面呈现局部正电荷,更易吸附带负电的CO₂分子,使CO₂R的起始电位比HER低0.2V,提升CO₂还原的选择性。
此外,层间限域效应也发挥作用,MXene层间距为0.7-0.9 nm时,CO₂分子的扩散与中间体的形成达到平衡,CH₄产率比单层体系高3倍。
这些计算不仅解析了MXene催化CO₂RR的路径特征与能垒分布,更建立了 “表面官能团–吸附强度–决速步能垒” 的关联模型,为设计高活性、高选择性的CO₂还原催化剂提供了理论指导,例如通过调控表面O官能团密度或引入N掺杂,可优化COOH吸附能,降低决速步能垒,推动MXene在碳循环与碳中和领域的应用。
DOI:10.1002/celc.202300598
经典案例:单原子 / MXene
以Gao等的《A theoretical study of electrocatalytic ammonia synthesis on single metal atom/MXene》为例,该研究通过系统的理论计算,揭示了单原子修饰 MXene 在电催化氮还原反应中的活性机制与应用潜力。
计算框架首先聚焦结构优化,构建Fe单原子负载于Ti₃C₂O₂的模型,通过DFT-PBE泛函优化几何结构,确定Fe原子的稳定吸附位点为Ti原子缺失形成的空位处,Fe与周围O原子形成配位键,键长约1.95Å,这种配位环境使Fe的d轨道处于适度杂化状态,为 N₂活化提供电子结构基础。
电子结构分析通过态密度(DOS)计算与Bader电荷分析展开,N₂分子在Fe位点的吸附能为-0.75eV,呈end-on吸附模式,Bader电荷计算显示0.32e从Fe转移至N₂的反键轨道,使N≡N键长从 1.10Å 增至1.13 Å,实现N₂的有效活化,电荷密度差分图直观显示了电子从Fe向N₂的转移路径。
NRR反应路径DFT计算构建了N₂逐步质子化生成NH₃的自由能台阶图,涵盖五步反应,每步均伴随电子转移与质子结合,计算结果显示N→*NNH步骤的自由能垒最高,为决速步,对应过电位0.84V,这一过电位低于传统Ru基催化剂,证实Fe/Ti₃C₂O₂的高活性。
理论指导意义体现在电子结构调控层面,计算发现Fe的d轨道与N₂的π*轨道杂化形成新的分子轨道,降低了N₂活化的能量势垒,这种 “单原子d轨道–底物反键轨道杂化” 机制为MXene载体设计提供了明确方向,即通过选择具有合适d电子构型的单原子与表面官能团,优化轨道匹配度,增强电子转移效率。
该研究首次建立了单原子 / MXene体系的NRR活性理论模型,其计算方法为后续其他单原子修饰MXene的催化研究提供了范式,推动了从理论设计到实验合成的转化,例如基于该理论指导合成的Fe/Ti₃C₂O₂催化剂,在0.1M Na₂SO₄电解液中NH₃产率达到15.6μg・h⁻¹・mg⁻¹,验证了理论预测的可靠性。
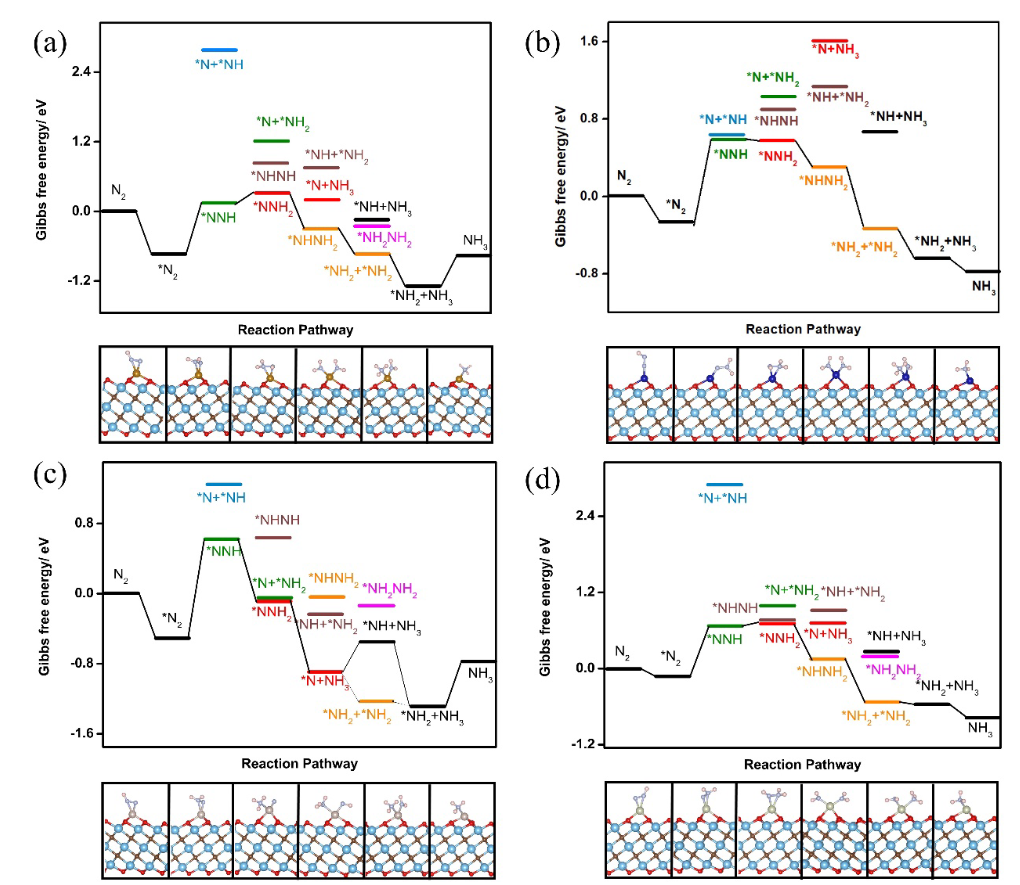
DOI:10.1016/S1872-2067(18)63197-3
总结
MXene电催化理论计算的发展通过多维度方法创新与跨尺度模型融合,构建了从基础研究到应用设计的完整理论体系。动态界面模拟聚焦固 – 液界面的真实反应环境,采用显式溶剂化模型研究质子转移与溶剂效应,例如在酸性HER中,AIMD模拟显示MXene表面的水合H₃O⁺离子通过氢键网络与表面O官能团作用,质子转移速率比真空模型计算快2个数量级,这种动态模拟弥补了静态DFT对溶剂化效应描述的不足,更准确反映实际反应条件下的界面行为。
机器学习加速设计基于图神经网络(GNN)等算法,以MXene的晶体结构、元素组成、表面官能团为输入特征,训练氢吸附自由能(ΔG_H*)预测模型,模型输出误差可控制在0.05eV以内,对1000种候选体系的筛选时间从传统DFT的数月缩短至数小时,实现了高效材料筛选;同时,通过特征重要性分析,识别出过渡金属电负性、表面官能团数量等关键描述符,为理性设计提供数据驱动的理论依据。
多尺度耦合模型则整合电子尺度的DFT计算、介观尺度的动力学蒙特卡洛(kMC)模拟与宏观反应器模型,例如在电解槽设计中,DFT计算的活性位点本征活性经kMC模拟扩展至微米尺度的反应动力学,再结合宏观传质模型,预测反应器的整体性能,这种跨尺度关联使理论计算更贴近工程应用需求。
新兴研究方向还包括强关联效应修正与极端条件模拟,进一步提升计算精度。这些进展不仅深化了对MXene催化机制的理解,更推动了其在能源转化领域的应用。未来,随着计算方法的持续优化与数据库的不断丰富,MXene理论计算将在 “原子级精准设计–宏量制备–器件集成” 链条中发挥更核心的作用,为开发高效、稳定、低成本的电催化剂提供全方位理论支撑。