

金属团簇颗粒的结构优化是材料科学和凝聚态物理中的一个重要研究方向。通过第一性原理计算方法,尤其是基于密度泛函理论(DFT)的VASP软件,可以对金属团簇的几何结构、电子结构和物理性质进行精确的理论模拟。以下将详细介绍如何使用VASP进行金属团簇颗粒的结构优化,并结合我搜索到的资料进行分析。
结构优化是通过计算确定材料在能量最小化状态下的几何构型。对于金属团簇而言,由于其尺寸较小,原子间的相互作用较强,因此结构优化尤为重要。结构优化的目标是找到团簇的基态结构,即能量最低的结构,从而能够准确预测其物理和化学性质。
VASP(Vienna Ab initio Simulation Package)是一种基于密度泛函理论的量子力学计算软件,广泛应用于材料科学、化学、物理等领域。它采用平面波基组和投影缀加波(PAW)方法,能够高效地处理原子、团簇、纳米材料、晶体等体系的电子结构和力学性质。VASP的结构优化功能是其核心功能之一,支持多种优化算法,如共轭梯度法(CG)、RMM-DIIS方法等。
1. 结构建模
在进行结构优化之前,需要构建团簇的初始结构模型。这可以通过多种方法实现,例如使用Vesta软件从CIF文件导入结构,或者通过MS软件手动构建结构。对于金属团簇,通常需要考虑其几何对称性和原子排列方式。例如,铝团簇(Alₙ)的结构可以通过实验或理论计算确定,然后作为初始结构进行优化。
2. 设置输入文件
在构建好初始结构后,需要生成相应的输入文件,包括POSCAR、INCAR、KPOINTS和POTCAR文件。POSCAR文件定义了团簇的原子种类、位置和晶格参数;INCAR文件设置了计算参数,如收敛标准、交换关联泛函、优化算法等;KPOINTS文件定义了布里渊区积分点;POTCAR文件提供了赝势信息。
POSCAR文件:定义了团簇的原子种类、位置和晶格参数。例如,对于Alₙ团簇,可以设置为一个二维或三维的超胞结构,以确保团簇之间的相互作用可以忽略。
INCAR文件:设置了计算参数,如收敛标准(EDIFF、EDIFFG)、优化算法(IBRION)、交换关联泛函(XC)、平面波截断能(ENCUT)等。例如,对于Alₙ团簇的优化,可以设置IBRION=2(共轭梯度法)和ISIF=2(仅离子弛豫)。
KPOINTS文件:定义了布里渊区积分点。对于非周期性分子或团簇,通常只需一个Γ点(1 1 1)即可。
POTCAR文件:提供了赝势信息,确保元素对应正确。例如,对于Alₙ团簇,可以使用Al的POTCAR文件。
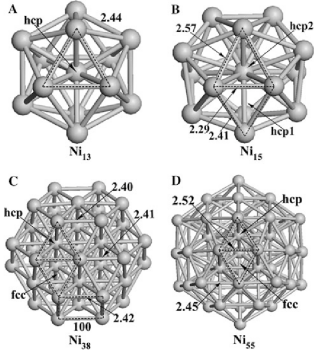

3. 结构优化计算
在设置好输入文件后,运行VASP进行结构优化计算。优化过程中,VASP会根据设置的收敛标准(如原子力小于0.05 eV/Å)和优化算法(如共轭梯度法)逐步调整原子位置,直到达到能量最小化状态。
优化算法:常用的优化算法包括共轭梯度法(CG)、RMM-DIIS方法和衰减分子动力学(DMD)。其中,RMM-DIIS方法在接近最小值时效果较好,而共轭梯度法适用于复杂问题。
收敛标准:通常设置原子力的收敛标准为0.01-0.05 eV/Å,能量的收敛标准为10⁻⁴ eV。
4. 结果分析
优化完成后,需要分析输出文件(如OUTCAR、CONTCAR、OSZICAR等),以确定团簇的基态结构和性质。例如,可以通过OUTCAR文件中的原子受力信息判断优化是否成功,如果个别原子受力过大,可能需要进一步优化。
基态结构:通过分析优化后的结构,可以确定团簇的稳定构型。例如,Alₙ团簇的稳定结构可能具有特定的几何对称性。
电子结构:通过计算团簇的态密度(DOS)和能带结构,可以分析其电子性质。例如,Alₙ团簇的磁矩可能在1 μ_B和2 μ_B之间变化。
1. 超原胞模型
对于金属团簇,由于其尺寸较小,通常需要使用超原胞模型来模拟其周期性边界条件。例如,Alₙ团簇可以放置在一个15 Å × 15 Å × 15 Å的超胞中,以确保团簇与其周期镜像之间的相互作用可以忽略。
2. k点选择
k点的选择对计算精度有重要影响。对于非周期性分子或团簇,通常只需一个Γ点(1 1 1)即可,而对于周期性体系,可以适当增加k点数量以提高计算精度。
3. 平面波截断能
平面波截断能(ENCUT)是影响计算精度的重要参数。通常建议将平面波截断能设置为系统中最大ENMAX值的1.3倍。
4. 自洽迭代
在结构优化过程中,需要进行自洽迭代,以确保电荷密度和势场的收敛。通常设置自洽迭代次数为100次以上。
1. Alₙ团簇的结构优化
以Alₙ团簇为例,研究表明其小团簇(n=2~7)具有磁性,磁矩在1 μ_B和2 μ_B之间变化。通过VASP进行结构优化,可以确定其稳定构型。例如,Al₈团簇的结合能随原子数增加而增大,但远小于铝晶体时的结合能。
2. Ptₙ团簇的结构优化
对于Ptₙ团簇,研究表明其基态结构通常为立体结构,对称性较低,多重度较高。通过VASP进行结构优化,可以确定其稳定构型。例如,Pt5Ni2团簇的稳定性最好,而Pt2Ni5的总磁矩最大。
3. W₃O₁₀团簇在TiO₂(110)表面的沉积构型优化
对于W₃O₁₀团簇在TiO₂(110)表面的沉积构型优化,研究表明其稳定结构取决于表面吸附位点和初始间距。通过VASP进行结构优化,可以确定其最佳吸附构型。
1. 方程–状态拟合
对于多孔材料(如MOFs),由于能量表面较为平坦,容易陷入局部最小值。此时,可以采用方程–状态拟合方法,通过固定体积优化,消除Pulay应力造成的误差。
2. 遗传算法结合DFT
对于复杂团簇体系,可以采用遗传算法结合DFT(GA-DFT)的方法,通过全局搜索找到最优结构。例如,GA-DFT方法已成功应用于多种键型的团簇体系,包括纯金属团簇、金属氧化物团簇、合金团簇等。
3. 机器学习辅助优化
近年来,机器学习技术也被引入到团簇结构优化中。通过训练模型预测团簇的稳定结构,可以显著提高计算效率。例如,基于密度泛函理论和机器学习的材料性能预测与优化研究中,利用机器学习算法对团簇的结构和性质进行预测和优化。
通过VASP进行金属团簇颗粒的结构优化,可以准确预测其几何结构、电子结构和物理性质。结构优化的关键在于合理设置输入文件、选择合适的优化算法和收敛标准,并结合实验数据进行验证。随着计算方法的不断发展,VASP在材料科学中的应用将更加广泛和深入。