

说明:双原子催化剂(DACs)通过物理吸附(范德华力)与化学吸附(轨道耦合、M-S键锚定)协同抑制多硫化锂(LiPSs)穿梭效应。DFT计算揭示转化路径:长链裂解(Li₂S₈→Li₂S₄能垒降至0.32 eV)和固相转化(Li₂S₂→Li₂S能垒0.18 eV)。
Fe-Co双原子体系通过d带中心调控与差分电荷转移实现“吸附–催化–解吸“循环,吸附能达-4.25 eV。未来结合机器学习与新型金属组合(如Mo-W),可加速高性能催化剂设计。
多硫化锂吸附-物理与化学双重调控
物理吸附
多硫化锂(LiPSs)的物理吸附机制主要依赖于材料表面与多硫化物分子间的范德华作用力,通过高比表面积结构(如石墨烯、碳纳米管)的空间限域效应抑制LiPSs扩散。
多孔碳材料凭借发达的孔道结构(孔径2-50 nm)与导电网络(电导率>100 S/cm)提供物理吸附位点,例如石墨烯的层间吸附能约-0.3 eV,可初步聚集Li₂S₄/Li₂S₆等中间产物,并通过导电基底加速电子传递以促进硫转化动力学。
然而,物理吸附强度有限(吸附能通常),无法完全阻断小分子Li₂S₂/Li₂S的穿梭效应,导致容量衰减。该机制虽能缓解但无法根治锂硫电池的循环稳定性问题,需结合化学吸附协同优化。
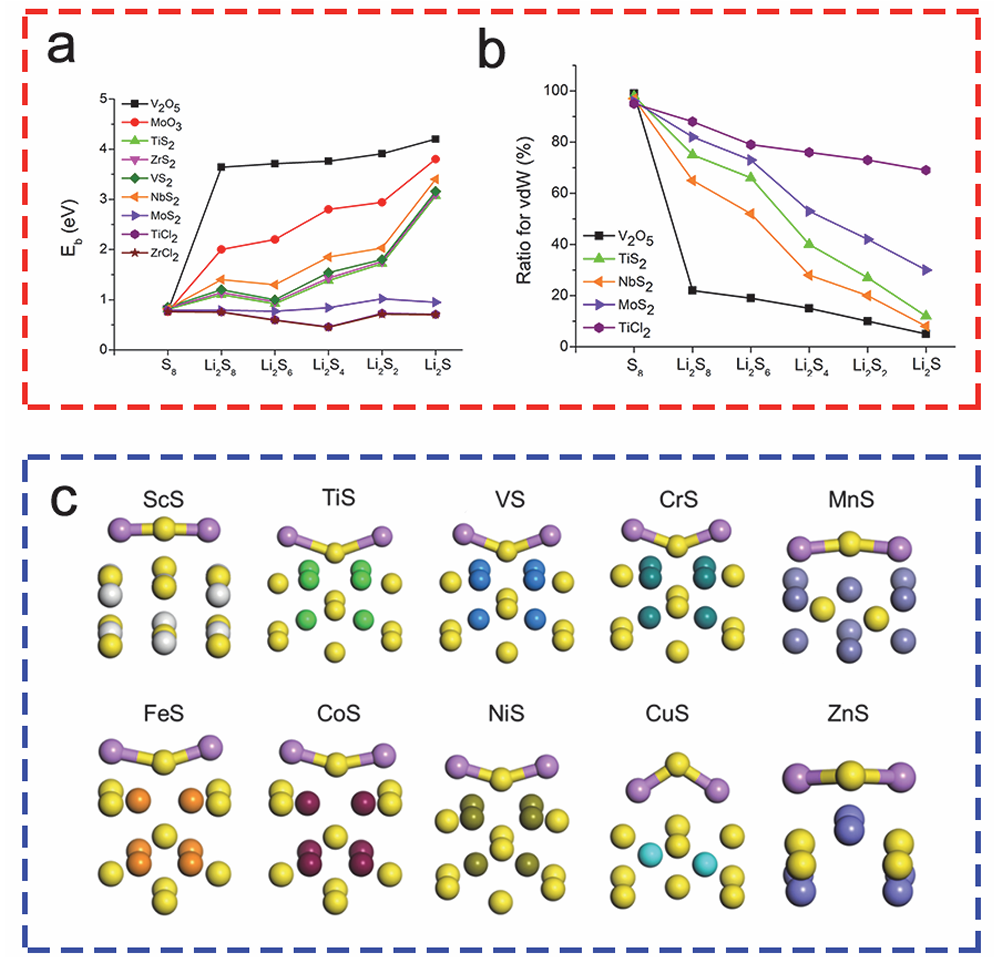

DOI:10.3866/PKU.WHXB202303061
化学吸附
多硫化锂的化学吸附机制通过轨道相互作用实现对 LiPSs 的化学键锚定,展现出物理与化学的双重调控智慧。
其中,非金属掺杂(如 N、O 等)凭借高电负性原子与 Li⁺的电荷转移过程,形成稳定的 Li–X 键,如同给多硫化锂戴上 “电荷枷锁”,使其在电极界面有序停留;
过渡金属活性位点(如 Fe、Co 等)则依靠金属 d 轨道与 S 原子的耦合效应,构建强韧的 M–S 键,例如 Fe₃C 对 Li₂S₆的吸附能可达 – 3.85 eV,同时通过拉长 S–S 键(键长增加)激活多硫化物裂解反应,如同用 “分子剪刀” 精准裁剪其结构。
这种化学吸附机制不仅通过化学键合力量实现对多硫化锂的定向捕获,还能通过电子轨道的协同作用调控其化学活性,为锂硫电池抑制穿梭效应、提升循环稳定性提供了兼具理论深度与应用潜力的解决方案,让纳米尺度的电荷与轨道相互作用成为优化电池性能的关键密码。
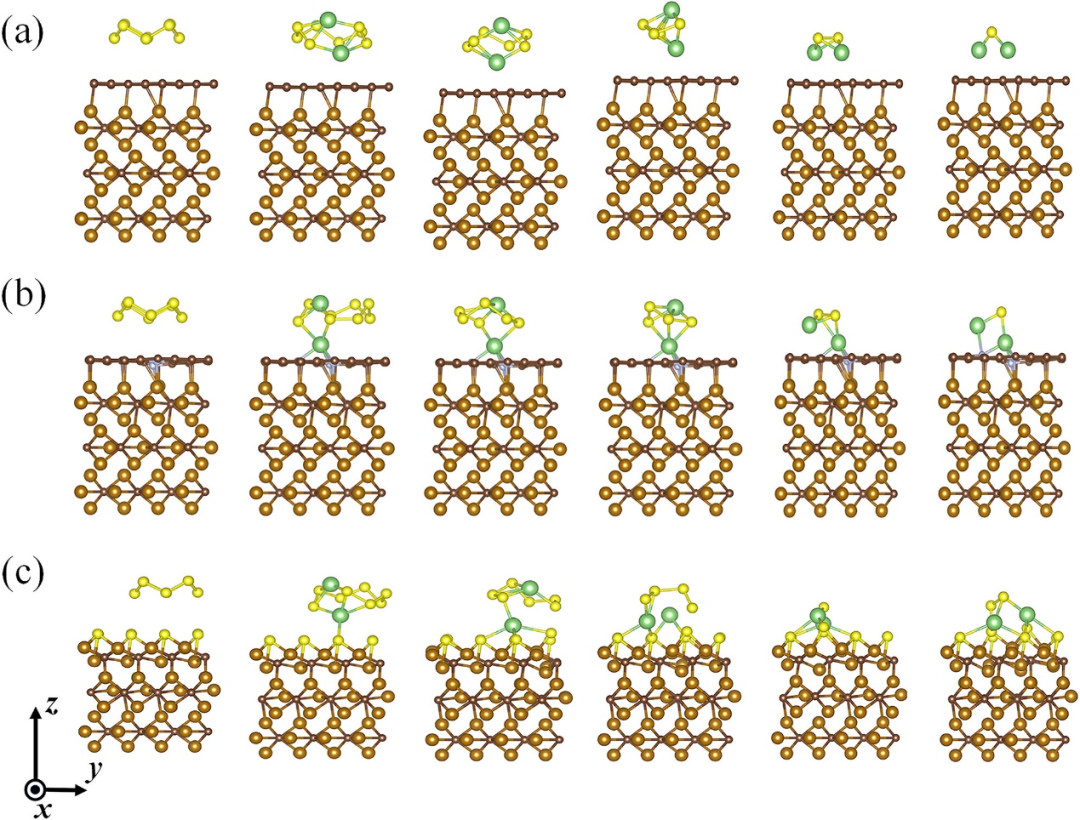
DOI:10.1021/acs.jpcc.3c04880
多硫化锂转化路径的DFT机理
LiPSs的转化涉及多个还原/氧化步骤,DFT计算通过吉布斯自由能台阶图揭示关键能垒:
长链→短链转化
多硫化锂(LiPSs)的转化路径可通过密度泛函理论(DFT)计算揭示其微观动力学奥秘,吉布斯自由能台阶图如同 “能量地图”,清晰展现长链向短链(如 Li₂S₈→Li₂S₄→Li₂S₂)转化过程中各还原 / 氧化步骤的关键能垒。
其中,吸附能作为“分子黏合剂” 发挥重要作用:强化学吸附可降低中间产物从催化剂表面脱离的概率,例如 LaNiO₃对 Li₂S₂的吸附能达 – 5.09 eV,显著高于 La₂O₃的 – 3.87 eV,凸显出高效锚定能力。
而 S–S 键活化则如同 “化学反应的启瓶器”,Fe₃C 纳米片通过将 Li₂S₆的 S–S 键长从 2.08 Å 拉长至 2.15 Å,削弱键能并促进断裂,从而加速长链碎片化进程。
这些 DFT 揭示的机理表明,催化剂可通过调控吸附能与键活化双重路径,优化 LiPSs 转化的能量壁垒与反应路径,为锂硫电池设计提供了 “从原子尺度加速反应动力学、抑制穿梭效应” 的科学依据,让微观层面的电子结构调控成为提升电池循环效率的核心钥匙。

DOI:10.1016/j.jechem.2023.03.046
固相转化
在多硫化锂(LiPSs)的固相转化阶段(如 Li₂S₂→Li₂S),密度泛函理论(DFT)计算通过吉布斯自由能台阶图,精准定位了制约固 – 液 – 固反应动力学的关键能垒节点。
以催化剂设计为例,CoP₂展现出独特的 “能量优化” 能力:其催化 Li₂S 分解的能垒仅为 1.08 eV,显著低于单金属催化剂 CoP 的 1.5 eV,这种能垒降低如同打通了化学反应的 “动力学高速路”,大幅加速固相产物的生成与分解循环。
从电子结构角度分析,双金属磷化物(如 CoP₂)通过调控金属 – 硫键的电子分布,降低了 Li₂S 成核与分解过程中的活化能,避免了单金属体系中常见的 “能垒瓶颈” 问题。
这种 DFT 揭示的机理表明,构建多元金属化合物可通过优化电子轨道耦合效率,精准调控固相转化的能量壁垒,为锂硫电池解决 “固态产物绝缘化” 难题提供了 “从原子级设计催化剂活性中心” 的新思路,让微观尺度的能垒调控成为提升电池充放电效率与循环稳定性的核心科学钥匙。
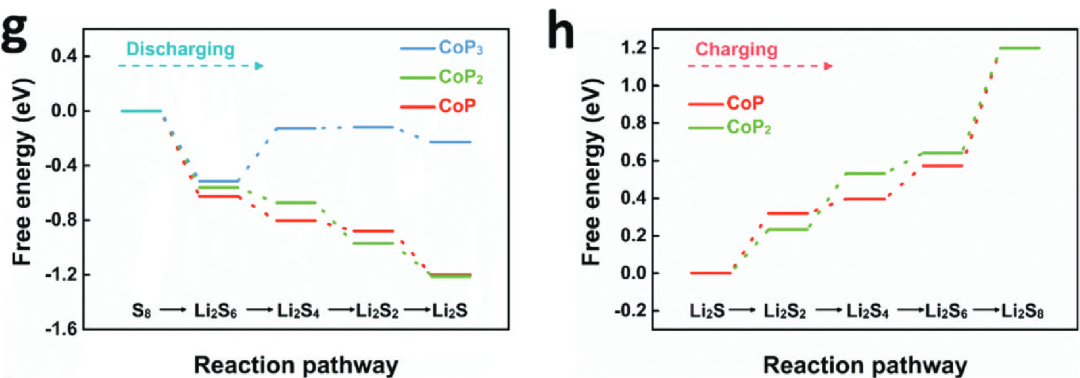
DOI:10.1002/aenm.202301551
双原子催化剂的协同效应
双原子催化剂(如 Fe-Co、Co-Ru 位点)通过电子结构与空间构型的协同设计,在锂硫电池多硫化锂(LiPSs)转化中展现出 “1+1>2” 的催化智慧。
从d 带中心调控来看,Fe-Co 双原子体系中两种金属的 d 带中心高度契合,犹如 “电子交响乐” 的精准协奏,使催化剂对 LiPSs 的吸附强度与解离能力达到动态平衡 —— 例如 Ni/Co-MOF@TCN 材料通过调整 d 带中心,将 Li₂S₆还原的吉布斯自由能垒降低 30%,显著优化反应动力学。
差分电荷分析进一步揭示,Fe-Co 双原子位点与 Li₂S₆间存在更强烈的电子转移(电子累积区域清晰显示界面相互作用增强),这种微观层面的电荷再分配为化学键的断裂与重组提供了能量支持。
在双向催化功能上,双原子位点构建了 “催化接力赛” 机制:放电过程中 Fe 位点作为 “还原引擎”,高效驱动长链 LiPSs(如 Li₂S₈)向短链(Li₂S₂)转化;充电时 Co 位点化身 “氧化助推器”,加速固态 Li₂S 分解为单质硫(S₈),避免活性物质因过度固化而失活。
这种基于电子耦合与空间协同的双功能设计,打破了单原子催化剂在反应路径调控上的局限性,为锂硫电池解决“穿梭效应” 与 “固态产物绝缘化” 难题提供了 “原子级精准协作” 的新思路 —— 当两种金属位点以特定电子态与几何构型协同作用时,不仅实现了对 LiPSs 吸附 – 解离 – 转化全流程的能量优化,更让催化剂在充放电循环中始终保持高效的双向催化活性,为高性能电池材料的设计掀开了 “双原子协同催化” 的崭新篇章。
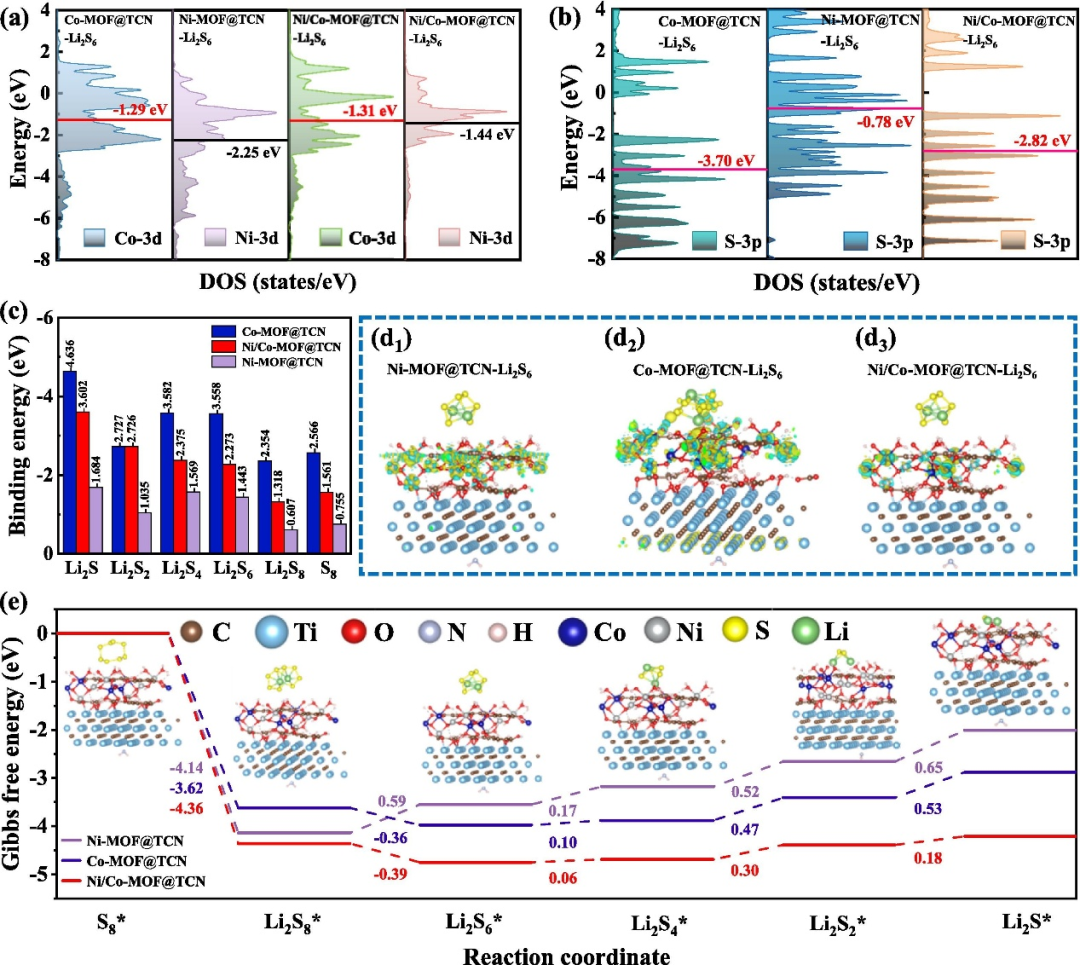
DOI:10.1016/j.cej.2025.160363
经典案例:Fe-Co双原子催化剂
以 Sun 等人(2023 年)关于 Fe-Co 双原子催化剂(DACs)的研究为例,该工作通过密度泛函理论(DFT)计算与实验验证,揭示了双原子体系在多硫化锂(LiPSs)转化中的独特优势,为锂硫电池催化机制研究提供了经典范本。
研究表明,Fe-Co DACs 对 Li₂S₆的吸附能达 – 4.25 eV,显著优于单原子 Fe 催化剂(Fe SACs,-3.82 eV)和单原子 Co 催化剂(Co SACs,-3.96 eV),这种更强的界面结合能力源于双原子间的电子协同效应 ——Fe 与 Co 通过电荷重分布形成 “双活性中心”,其中 Fe–S 键如同 “分子锚” 优先捕获长链 LiPSs(如 Li₂S₈),而 Co–S 键则充当 “化学剪刀” 加速短链中间体(如 Li₂S₂)的解离,形成 “吸附 – 催化 – 解吸” 的高效循环。
吉布斯自由能台阶图进一步量化了这种协同效应的动力学优势:在长链碎片化路径中,Li₂S₈→Li₂S₆的能垒从单原子催化剂的 0.58 eV 降至 0.45 eV,Li₂S₆→Li₂S₄的能垒从 0.49 eV 降至 0.32 eV,反应活化能的显著降低表明双原子位点可大幅加速 LiPSs 的链长缩减进程;
在关键的固相转化阶段(Li₂S₂→Li₂S),Fe-Co DACs 的能垒仅为 0.18 eV,仅为单原子催化剂的 1/3,这意味着其能有效抑制固态 Li₂S 的绝缘化沉积,提升电池充放电效率。
从电子结构角度分析,Fe 与 Co 原子的 d 轨道通过轨道杂化形成独特的电子云分布,使双原子位点既能通过 Fe 位点的高电负性实现对长链 LiPSs 的强吸附,又能利用 Co 位点的 d 带中心调控优化短链中间体的解离路径。
这种“双功能分工” 打破了单原子催化剂在吸附强度与催化活性间的权衡限制—— 单原子体系常因吸附过强导致中间体滞留或吸附过弱导致穿梭效应,而 Fe-Co DACs 通过原子级精准设计,在界面相互作用与反应动力学之间找到了理想平衡点。
该研究不仅通过详实的计算数据揭示了双原子催化剂的“1+1>2” 机制,更展现了从原子尺度设计高效催化位点的科学方法论。
当 Fe 与 Co 以特定间距与电子态协同作用时,其构建的双活性中心如同 “纳米级催化工厂”,在锂硫电池的充放电循环中同步实现了对 LiPSs 转化全流程的能量优化,为解决锂硫电池的 “穿梭效应” 与 “固态产物动力学瓶颈” 提供了兼具理论深度与工程潜力的解决方案,也为后续双金属催化体系的设计提供了可借鉴的 “原子级协同模板”。
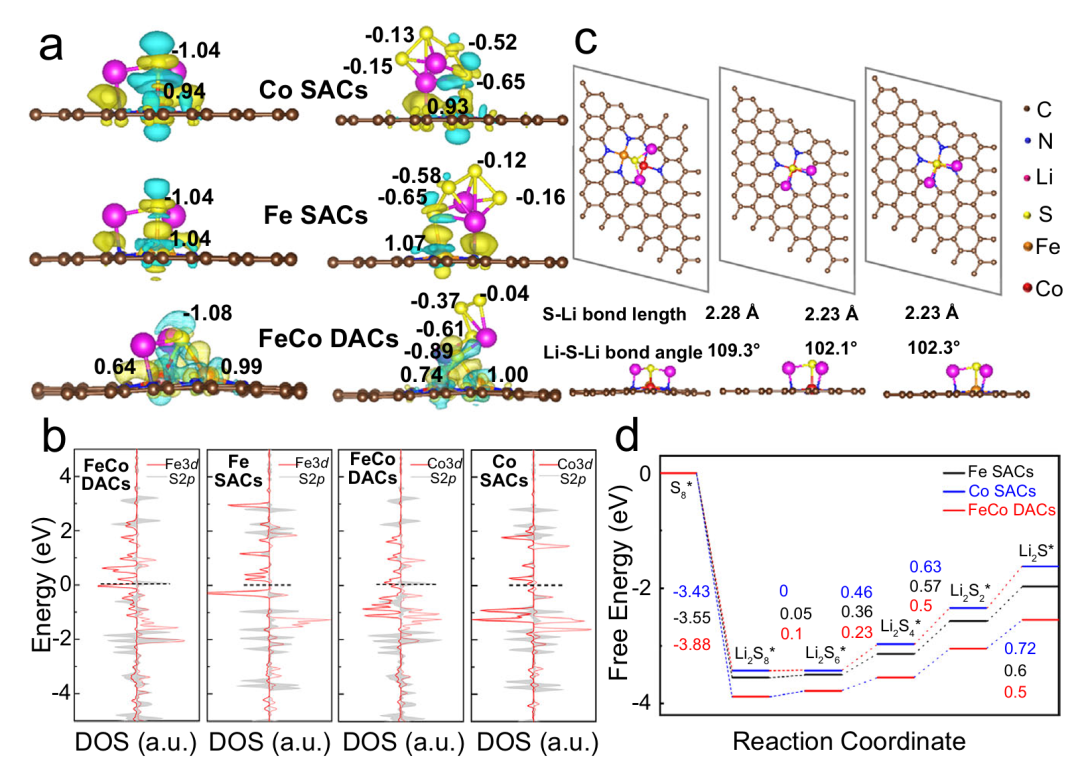
DOI:10.1038/s41467-022-35736-x
总结
双原子催化剂通过多活性位点协同,显著优化了 LiPSs 的吸附与转化路径,为锂硫电池性能提升奠定了原子级调控基础。
未来研究将从三方面展开:一是开发更精准的 d 带中心调控策略,通过电子结构微调平衡对 LiPSs 的吸附强度与解吸效率,避免过强吸附导致的中间体滞留或过弱吸附引发的穿梭效应;
二是探索 Mo-W、Mn-Ni 等新型双金属组合,利用不同金属的电子特性与轨道匹配性拓展催化功能多样性,满足多阶段反应的差异化需求;三是结合机器学习与 DFT 计算,构建高效的催化剂筛选模型,如同为材料设计配备 “智能加速器”,大幅缩短高性能催化剂的研发周期。
随着对 DFT 台阶图揭示的反应路径与能垒机制的深入解析,锂硫电池正逐步突破 “理论 – 实验” 转化瓶颈,这些研究方向的推进将为电池实用化进程注入新动能,让原子尺度的协同催化智慧真正转化为能源领域的技术突破。