


分子轨道理论(MO理论)是一种用于描述分子中电子分布与化学键的理论模型,它认为分子中的电子并非局限于特定的原子轨道,而是分布在整个分子范围内的分子轨道中。
该理论通过原子轨道的线性组合生成分子轨道,进而解释了分子结构、化学键的性质、电子分布、磁性等现象。分子轨道理论不仅能帮助我们理解分子的稳定性和反应性,还能在分子的光谱特性和磁性研究中提供深刻的洞见。
分子轨道理论详解
分子轨道理论(Molecular Orbital Theory,简称MO)是量子化学中用于描述化学键形成和分子电子结构的核心理论之一。它通过构建分子整体的电子波函数,揭示分子中电子的分布与能量状态。以下将从多个维度展开详细论述
基本定义与核心概念
分子轨道的本质
分子轨道是描述分子中单个电子运动状态的波函数。它可以通过原子轨道的线性组合(LCAO-MO)或杂化形成。例如,氢分子(H₂)的σ成键轨道是由两个氢原子的1s轨道同相位叠加而成的,而σ*反键轨道则是由异相位叠加形成的。这种叠加方式决定了电子在分子中的分布和能量状态,从而影响分子的化学性质和稳定性。
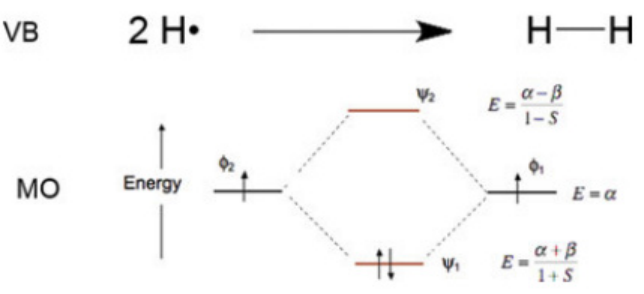
轨道分类与能量关系
成键轨道(Bonding MO):成键轨道的能量低于原始原子轨道,电子密度主要集中在原子核之间的区域。这种轨道的电子分布有助于促进化学键的形成,因为电子在核间区域的高密度可以降低系统的能量,从而增强原子之间的结合力。例如,在氢分子(H₂)中,σ成键轨道的电子密度主要集中在两个氢原子核之间,使得两个氢原子能够紧密结合在一起,形成稳定的分子。
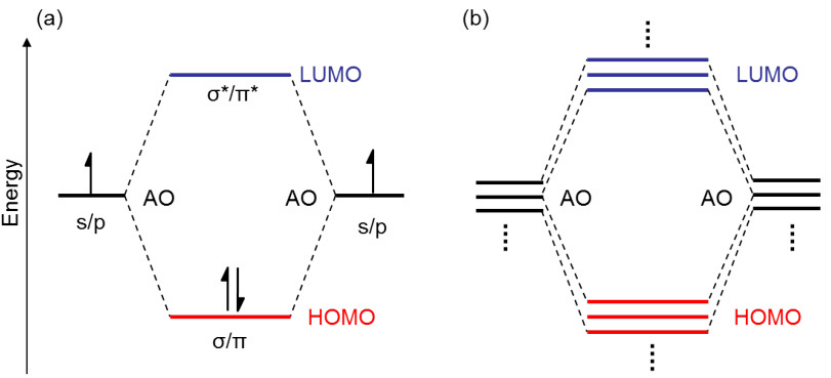
反键轨道(Antibonding MO):反键轨道的能量高于原子轨道,在原子核之间存在电子密度节点。这意味着电子在核间区域的密度较低,反而会削弱原子之间的键合。例如,σ*反键轨道的存在会使得分子的稳定性降低,因为电子在该轨道上的分布会抵消部分成键轨道的稳定作用。在一些分子中,反键轨道的电子填充情况会影响分子的化学反应活性和稳定性。
非键轨道(Nonbonding MO):非键轨道的能量接近原子轨道,电子主要分布在原子自身的周围,不参与成键。这种轨道通常由孤对电子占据,例如在水分子(H₂O)中,氧原子的两对孤对电子占据非键轨道,它们对分子的化学键形成没有直接贡献,但会影响分子的几何构型和某些化学性质。
轨道填充规则
能量最低原理:电子会优先填充能量较低的轨道,因为这样可以使分子的能量处于最低状态,从而达到最稳定的结构。例如,在氧分子(O₂)中,分子轨道能级图显示π轨道的能量较高,因此电子在填充轨道时会先填满能量较低的轨道,最后才会填充到π轨道。由于π*轨道的高能级,氧分子的基态存在两个单电子,这使得氧分子具有顺磁性。。
泡利不相容原理:每个分子轨道最多只能容纳两个自旋相反的电子。这一原理保证了电子在轨道中的排布不会出现违反量子力学规则的情况,同时也决定了分子轨道中电子的填充顺序和自旋状态。例如,在氮分子(N₂)中,每个成键轨道和反键轨道都按照泡利不相容原理填充电子,使得氮分子的电子排布符合其稳定的三键结构。
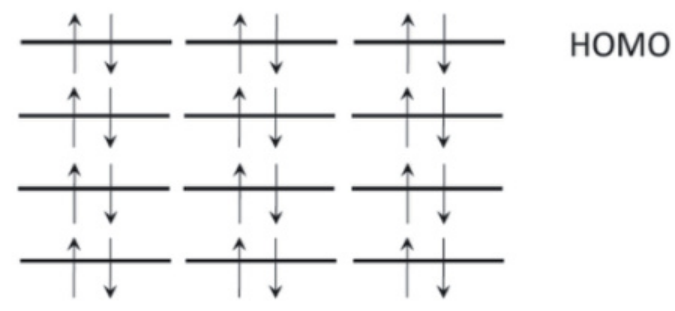
洪特规则:在简并轨道(即能量相同的轨道)中,电子会优先分占不同的轨道,并且自旋平行。这一规则有助于最大化电子之间的交换能,从而降低分子的能量。例如,在氧分子(O₂)的π轨道中,两个电子分别占据两个简并的π轨道,并且自旋平行,这使得氧分子的电子排布更加稳定。
HOMO与LUMO
最高占据分子轨道(HOMO):HOMO是指在分子中电子占据的最高能量轨道。它决定了分子的电子给予能力,即分子在化学反应中作为亲核试剂的活性。例如,在一些有机反应中,分子的HOMO电子可以被吸引到电正性中心,从而发生亲核进攻。HOMO的能量越高,分子越容易给出电子,亲核性越强。
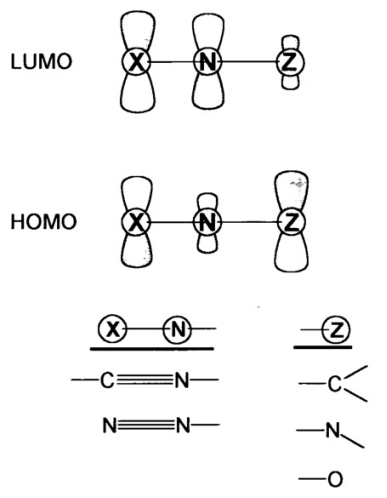
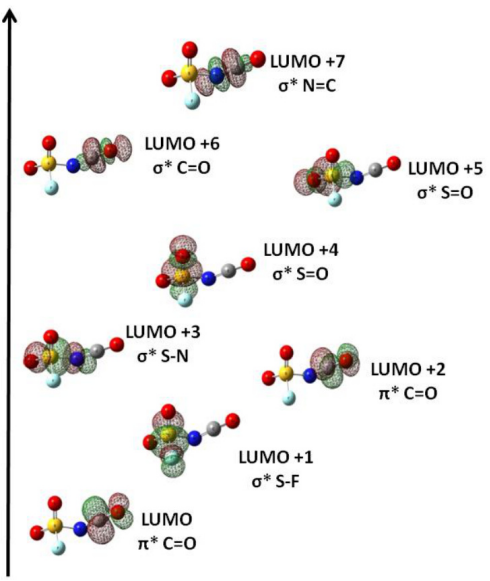
最低未占据分子轨道(LUMO) :LUMO是指在分子中尚未被电子占据的最低能量轨道。它决定了分子的电子接受能力,即分子在化学反应中作为亲电试剂的活性。例如,在亲电加成反应中,亲电试剂的电子可以被吸引到分子的LUMO中,从而发生反应。LUMO的能量越低,分子越容易接受电子,亲电性越强。
HOMO与LUMO的能级差(带隙):HOMO与LUMO之间的能级差被称为带隙,它对分子的光学和电学性质有着重要影响。例如,在半导体材料中,带隙的大小决定了材料的导电性和光学吸收特性。较小的带隙意味着电子更容易从HOMO跃迁到LUMO,从而使材料具有更好的导电性和光吸收能力。
不同分子的能级差
发展历史与关键人物
奠基者与里程碑
弗里德里希·洪特(1928):洪特是分子轨道理论的奠基人之一,他首次提出了分子轨道的概念,并成功解释了氧分子(O₂)的顺磁性。这一发现为分子轨道理论的发展奠定了基础,使得人们开始认识到分子轨道在描述分子电子结构和化学性质方面的重要性。
罗伯特·马利肯(1932):马利肯对分子轨道理论进行了系统化的发展,提出了分子轨道能级图,为分子轨道理论的应用提供了重要的工具。他的工作使得分子轨道理论能够更加直观地描述分子中电子的分布和能量状态。由于他在量子化学领域的杰出贡献,马利肯荣获了1966年的诺贝尔化学奖。。
约翰·兰纳–琼斯:兰纳–琼斯在分子轨道理论的发展中也发挥了重要作用,他发展了原子轨道的线性组合(LCAO-MO)方法,使得分子轨道能够通过数学方法进行定量描述。这一方法为分子轨道理论的计算和应用提供了坚实的基础。
理论演变
早期应用:在分子轨道理论的早期发展阶段,它主要用于定性解释双原子分子的光谱现象。例如,通过分子轨道理论可以很好地解释氧分子(O₂)和氮分子(N₂)的光谱特性,以及它们的化学键性质和稳定性。这些早期的应用为分子轨道理论的进一步发展积累了宝贵的经验。
数学框架完善:1951年,C. C. J. Roothaan建立了哈特里–福克方程的自洽场(SCF)方法。这一方法的出现极大地完善了分子轨道理论的数学框架,使得分子轨道理论能够更加精确地描述分子的电子结构和能量状态。自洽场方法通过迭代计算,逐步优化分子轨道的波函数,从而得到更加准确的结果。
计算化学发展:随着计算化学的不断发展,分子轨道理论与现代计算方法相结合,取得了显著的进展。例如,密度泛函理论(DFT)的出现为分子轨道理论的计算提供了更加高效的方法。DFT通过引入交换–相关势,能够更加准确地描述电子之间的相互作用,从而提高基态能量的计算精度。
此外,基组的不断改进(如STO-3G、6-31G等)也为复杂分子的计算提供了更好的基础,使得分子轨道理论在研究大型分子和复杂体系时变得更加可行。
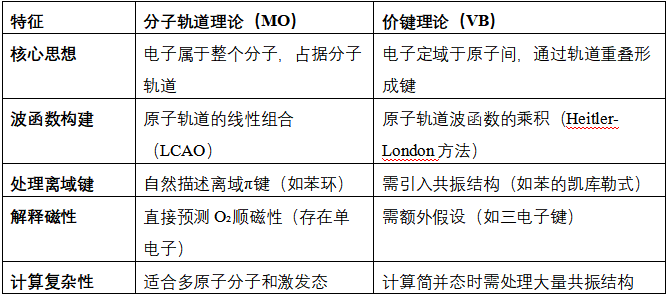
分子轨道与价键理论的区别
应用案例
简单分子(H₂、O₂)
H₂:两个1s轨道形成σ(成键)和σ*(反键)轨道,基态电子填满σ轨道,键级为1。
O₂:分子轨道能级图显示存在两个单电子占据π*轨道,导致顺磁性和键级2。
有机化合物
乙烯(C₂H₄) :sp²杂化形成σ键,未杂化p轨道侧向重叠形成π键。
苯(C₆H₆) :离域π轨道形成环状共轭体系,降低分子能量。
乙炔(C₂H₂) :sp杂化形成σ键,两对p轨道形成正交π键,构成三键。
配位化合物
σ-供体与π-受体:CO中孤对电子向金属σ轨道供电子(σ键),同时金属d轨道向CO π*轨道反馈(π键)。
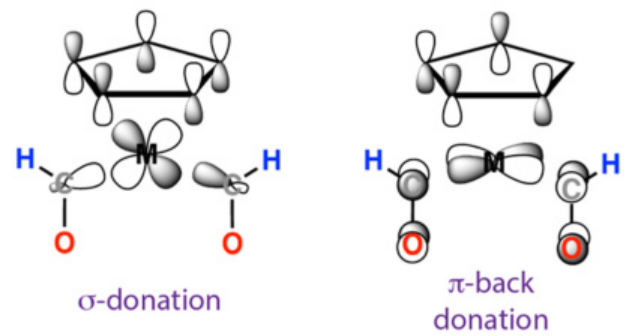
磁性团簇:分子轨道理论解释高自旋与低自旋配合物的电子排布差异。
有机太阳能电池(OSCs):基于 HOMO/LUMO 能级匹配的分子设计
有机太阳能电池依赖施主(D)– 受主(A)异质结界面的电荷分离,其效率直接取决于施主材料的最高占据分子轨道(HOMO)与受主材料的最低未占据分子轨道(LUMO)能级匹配度。分子轨道理论通过精确计算能级差(EHOMO,D−ELUMO,A),指导高效给受体材料的设计与筛选。
经典体系:富勒烯衍生物与聚合物的能级调控
PCBM([6,6]- 苯基 – C61 – 丁酸甲酯):作为传统受主材料,其 LUMO 能级约为 – 4.3 eV,需匹配施主聚合物的 HOMO 能级(如 P3HT 的 HOMO 为 – 5.1 eV),形成 0.8 eV 的驱动力以确保激子分离。分子轨道计算显示,富勒烯笼的 π* 轨道离域性增强了电子接受能力,但刚性结构限制了与聚合物的界面相容性。
非富勒烯受主(NFAs)的突破:以 Y6 为例,其 LUMO 能级通过氟原子取代调控至 – 3.8 eV,与施主材料 PM6 的 HOMO 能级(-5.2 eV)形成 1.4 eV 的能级差,结合分子轨道离域的苯并二噻吩单元,实现了 18% 以上的能量转换效率。计算表明,Y6 的 LUMO 主要分布在缺电子的噻吩 -[3,4-c] 吡咯 – 4,6 – 二酮(TPD)单元,有利于电子快速传输至电极。
新型给体材料:共轭小分子的轨道工程
D-A-D 型分子(如 BDT-4Cl):通过引入吸电子基团(如 – Cl)降低 HOMO 能级(从 – 5.0 eV 降至 – 5.3 eV),同时扩展 π 共轭体系(如联噻吩单元)降低 LUMO 能级,减小带隙至 1.6 eV,增强可见光吸收。分子轨道模拟显示,氯原子的强吸电子诱导效应使 HOMO/LUMO 能级整体下移,同时通过空间位阻调控分子堆积方式,促进电荷传输路径的形成。
杂原子掺杂(N/S/O):在共轭骨架中引入氮原子(如吩嗪单元),其孤对电子与π 体系共轭,使 HOMO 能级升高 0.2 eV,而硫原子的 d 轨道参与共轭可降低 LUMO 能级 0.1 eV,通过精确调节实现与 NFAs 的能级精准匹配。
二维半导体材料:分子轨道离域性与能带结构调控
二维材料(如过渡金属二硫族化合物 TMDs、石墨烯衍生物)的电子性质由层内分子轨道的离域程度决定,分子轨道理论通过分析轨道重叠积分与能带色散关系,指导材料的能带工程与缺陷调控。
二硫化钼(MoS₂):从间接带隙到直接带隙的转变
块体 MoS₂:层间通过弱范德华力堆叠,层内 Mo 原子的 d 轨道与 S 原子的 p 轨道形成 σ 键(成键轨道)和 σ* 反键轨道,价带顶(VBM)与导带底(CBM)位于布里渊区不同点(K 点与 Γ 点),属于间接带隙半导体(1.2 eV),光吸收效率低。
单层 MoS₂:量子限域效应导致轨道离域性改变,VBM 与 CBM 均移至 K 点,转变为直接带隙(1.8 eV)。分子轨道计算表明,单层中 Mo 的 dxy 轨道与 S 的 px/py 轨道形成强 π 共轭,轨道重叠积分从块体的 0.15 增大至 0.32,增强了光跃迁概率,使其成为高效光催化剂与光电探测器材料。
石墨烯衍生物:缺陷工程调控轨道杂化
氧化石墨烯(GO):引入环氧基团(-O-)与羟基(-OH)导致 sp² 杂化碳转化为 sp³ 杂化,局域化的 σ 轨道在价带顶上方形成缺陷态(HOMO 能级升高),带隙从 0 eV(石墨烯)打开至 2.5 eV,适用于紫外光探测。分子轨道模拟显示,羟基的孤对电子占据非键轨道(n 轨道),与相邻碳原子的 p 轨道形成弱共轭,导致光生载流子寿命缩短至纳秒级。
氮掺杂石墨烯(NG):吡啶型氮原子替代碳原子引入孤对电子(占据 sp² 杂化轨道,HOMO 能级升高 0.3 eV),而石墨型氮的 p 轨道与石墨烯 π 体系共轭(LUMO 能级降低 0.2 eV),带隙调控至 0.8 eV,同时形成高效氧还原反应(ORR)活性位点,其轨道重叠积分比纯石墨烯提高 40%,增强了电子转移效率。
模型构建到实验验证
第一性原理计算流程
结构优化:利用 DFT(如 VASP 软件)优化分子几何结构,计算 HOMO/LUMO 能级、轨道分布与能带结构。例如,在设计新型有机发光材料时,通过 B3LYP/6-31G计算预测分子的前线轨道分布,判断发光机制(如π-π跃迁或电荷转移跃迁)。
激发态模拟:采用含时密度泛函理论(TD-DFT)计算 LUMO+1、LUMO+2 等激发态轨道,预测紫外 – 可见吸收光谱与荧光发射波长。如钙钛矿材料 MAPbI₃的激子结合能(约 50 meV)可通过分子轨道的电子 – 空穴波函数重叠积分计算得出。
电荷传输模拟:基于分子轨道的重叠矩阵,使用 Nevanlinna-Pick 定理计算空穴 / 电子迁移率。例如,在有机场效应晶体管(OFET)中,共轭分子的 π 轨道面内重叠程度(如并五苯的轨道重叠积分 > 0.2)决定了迁移率可达 1 cm²/Vs 以上。
实验验证手段
光电子能谱(UPS/XPS):直接测量 HOMO 能级(UPS 结合能)与 LUMO 能级(通过紫外吸收边估算),如对酞菁铜(CuPc)的 UPS 测试显示其 HOMO 能级为 – 5.2 eV,与 DFT 计算值(-5.1 eV)吻合。
空间分辨光谱:利用扫描隧道显微镜(STM)成像分子轨道分布,如在石墨烯边缘观测到局域化的 π 轨道态密度,验证边缘缺陷对轨道离域性的影响。
器件性能测试:通过光伏器件的短路电流(Jsc)与开路电压(Voc)反推能级匹配度,如 Voc∝(EHOMO,D−ELUMO,A)−0.3eV(经验公式),指导给受体材料的二次筛选。
总结
分子轨道理论通过量子力学框架统一描述化学键的本质,成功解释了从简单双原子分子到复杂共轭体系的电子行为。其核心优势在于自然处理离域性与激发态,但计算挑战促使理论与计算方法的持续革新。结合现代计算工具,分子轨道理论在材料设计、催化机理和光谱解析等领域仍具有不可替代的价值。