

差分电荷密度
差分电荷密度(Charge Density Difference, CDD)是量化电子结构变化的重要工具,通过计算体系相互作用前后的电荷密度差异(Δρ = ρAB − ρA − ρB),直观揭示电荷的重新分布。其核心思想是消除孤立组分的贡献,突出键合或吸附等过程中电子的转移与重组。
差分结果中,正值区域(如蓝色等值面)表示电子积累,负值区域(如红色等值面)表示电子耗散,二者空间分布直接反映电荷转移方向与程度。
为什么可以分析电荷转移
电荷密度差分分析电荷转移的物理本质源于电子云重构的可视化与定量化。当两个组分(如分子与表面)接近时,电子会因电负性差异、轨道杂化或静电作用发生迁移。差分图通过对比复合体系与孤立体系的电荷密度,剥离了未相互作用时的本底电子分布,仅保留由化学键或物理吸附诱导的电子再分配。
例如,若差分图显示A组分区域电子耗散、B组分区域电子积累,则表明电荷从A向B转移。因此,CDD兼具直观性与定量性,成为研究界面电荷转移、催化活性位点或分子极化的关键手段。
应用
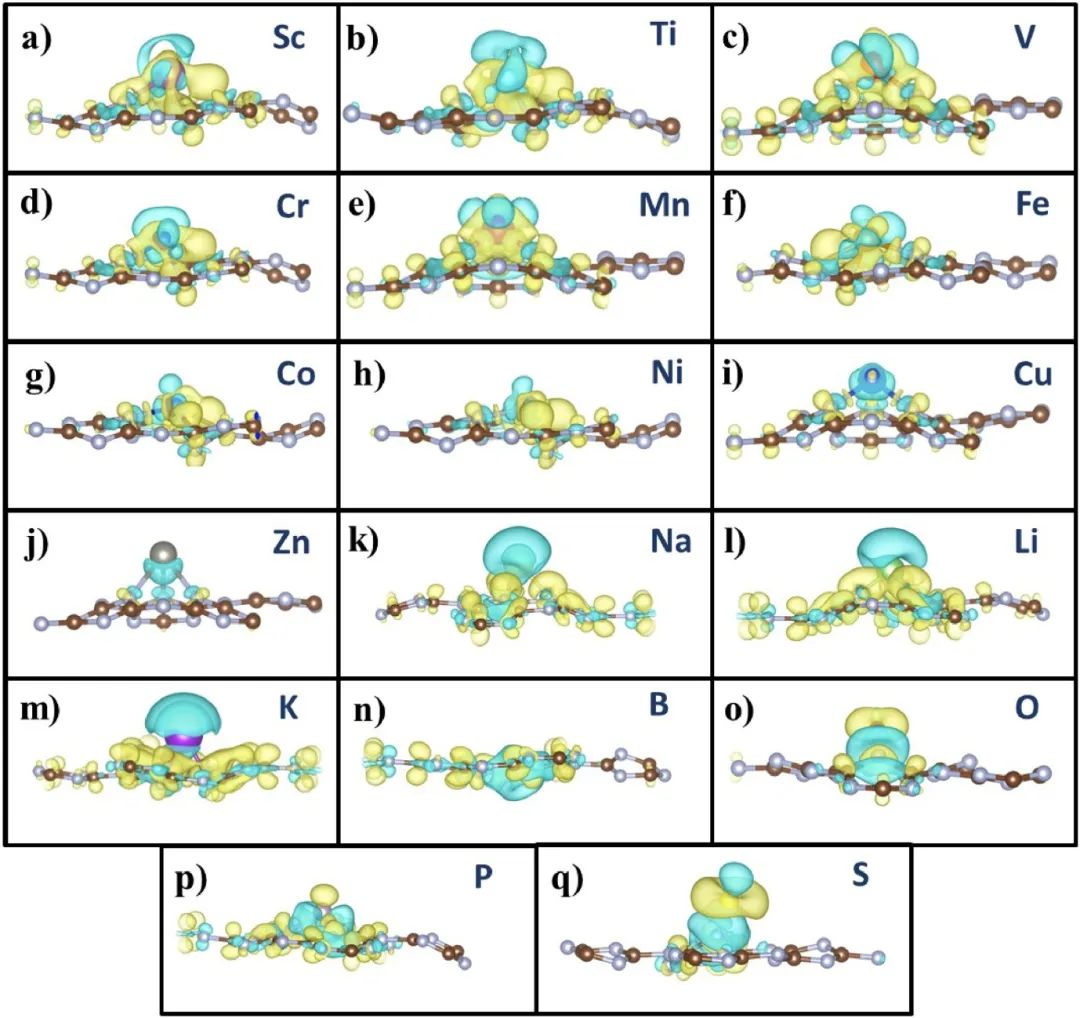
通过差分电荷密度图可以清晰展示不同金属(TM/AM/NM)掺杂gC₃N₄后的电子转移机制和成键特性。对于3d过渡金属(Sc至Ni,图a-h),差分图中黄绿交替区域表明其部分填充的d轨道与gC₃N₄中N的p轨道发生杂化,形成成键(电荷积累,黄色)与反键(电荷耗散,绿色)态共存的现象,揭示了共价相互作用的复杂性。
例如,Fe、Co等因d电子未填满,电荷在金属与基底间双向流动,体现强轨道耦合。而Cu、Zn(图i-j)因d轨道全充满,仅显示绿色耗散区,说明电子从金属单向转移到gC₃N₄,符合其弱还原性。
碱金属(Na、Li、K,图k-m)因电负性极低,绿色耗散区集中在原子周围,证实电子完全离域到基底,形成离子键特征。非金属中,B(图n)在键合区积累电荷,显示共价键形成;O(图o)因高电负性吸引电子,导致自身周围强积累而键合区耗散,呈现极性键;S和P(图p-q)则因中等电负性同时存在积累与耗散,反映混合键合特性。
整体上,差分图不仅验证了电负性差异驱动的电荷转移方向,还通过空间分布揭示了键合类型(离子/共价/极性),为理解掺杂原子与gC₃N₄的相互作用机制提供了直观电子尺度证据。

Bader 电荷等电荷布局
Bader电荷是基于Bader提出的“原子中的原子”(Atoms in Molecules, AIM)理论计算的一种电荷划分方法。该理论通过分析电子密度的拓扑结构(如临界点、梯度路径等),将分子或固体中的电荷密度分布划分为不同的原子区域(Bader体积),并通过积分每个区域内的电子密度来确定原子的净电荷。Bader电荷的计算公式为:
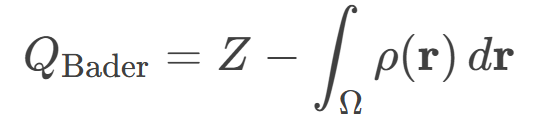
其中, Z 为原子核电荷数,Ω为Bader体积,ρ(r)为电子密度。Bader电荷的正负和大小反映了原子在化学环境中的电子得失情况,是量化电荷转移的重要指标。
为什么可以分析电荷转移
Bader电荷能够精确分析电荷转移,因为它基于电子密度的物理划分,避免了传统布居分析(如Mulliken或Hirshfeld)中基组依赖性强或划分方式主观的缺点。在界面或分子体系中,Bader电荷可以直接比较相互作用前后原子的电荷变化,从而明确电子流动方向。
例如,若金属原子在吸附后的Bader电荷显著增加(正值增大),说明其失去电子;若基底原子的Bader电荷减少(负值增大),则表明其获得电子。此外,Bader电荷的空间分布可揭示局域电荷转移(如化学键形成)或长程电荷重排(如金属-半导体接触的电荷再分配),使其成为研究催化、吸附、掺杂等过程中电子行为的关键工具。
此外,Bader电荷的定量化使其适用于不同体系间的比较。例如,在催化研究中,可通过比较不同金属吸附质的Bader电荷变化,判断其电子给体/受体能力;在半导体异质结中,Bader电荷可揭示界面处的电荷积累或耗尽,解释能带弯曲的起源。
结合差分电荷密度分析,Bader电荷不仅能确定电荷转移的净量,还能定位电子转移的空间分布,从而全面解析化学键的本质(共价、离子或金属键)。因此,Bader电荷是理论计算和材料模拟中不可或缺的工具,为理解电子结构调控和化学反应机制提供了坚实的理论基础。
应用
Bader电荷分析揭示了Fe,N₃-GDY催化剂在氢吸附过程中的电荷转移机制及其对HER活性的影响。在H1和H3两种吸附位点中,Fe原子均表现为电子受体(H1: +0.799 e;H3: +0.728 e),而氮原子(N)作为电子供体(H1: -1.162 e;H3: -1.653 e),表明Fe-N协同作用通过电荷再分配优化了催化活性。
碳原子(C1/C2)的Bader电荷变化尤为关键:H1位点吸附时,C1和C2电荷减少(+0.229 e/+0.188 e),反映其与周围原子的相互作用减弱;而在H3位点,电荷显著增加(C1: +0.460 e;C2: +0.292 e),说明碳原子通过电子积累稳定了吸附氢(H),这与H3位点更优的HER性能(ΔGH = 0.020 eV)直接相关。
此外,H*本身带轻微正电(+0.104~+0.108 e),表明其与基底间存在极化作用,电荷密度差分图(CDD)进一步证实电子从Fe/N向H-C键区域转移,形成共价相互作用。
功函数
功函数(Work Function)是指将一个电子从固体内部刚刚移到表面所需克服的最小能量,它等于真空能级与固体费米能级之间的能量差,数学表达式为:
φ=Evac-Efer
其中,φ 表示功函数,Evac 是真空能级,Efer 是固体的费米能级 。真空能级是电子在真空中具有的能量,而费米能级是固体中电子的化学势,反映了电子在固体中的填充水平 。
为什么可以分析电荷转移
当两种不同材料相互接触或发生化学反应时,由于它们的功函数不同,会导致电子在界面处发生转移,以达到新的电子平衡状态 。功函数较低的材料会失去电子,功函数较高的材料会获得电子,直到两者的费米能级对齐 。
通过测量或计算材料的功函数,并分析其在不同条件下(如吸附、掺杂等)的变化,可以推断出材料表面或界面处的电荷转移情况 。例如,当金属与半导体接触时,根据它们的功函数差异可以判断电子的转移方向,进而确定接触形成的是肖特基势垒还是欧姆接触 。
应用
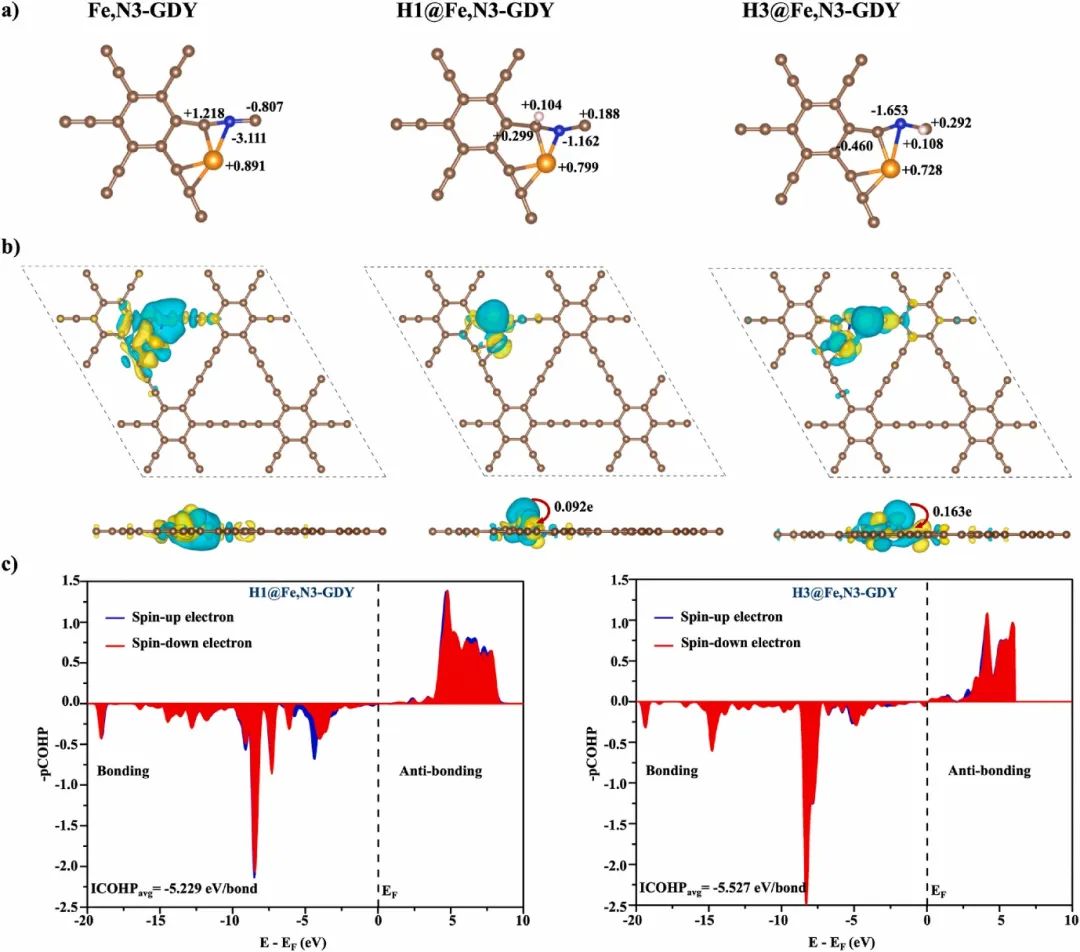
在接触前(图a),p型半导体BiOBr的费米能级靠近价带(VB,0.24 eV),而n型ZnWO₄的靠近导带(CB,0.18 eV),两者存在显著能级错位(BiOBr的CB为3.10 eV,ZnWO₄为3.44 eV)。
接触后(图b),由于费米能级平衡需求,电子从高能级(ZnWO₄的EF)向低能级(BiOBr的EF)流动,导致BiOBr的能带上移、ZnWO₄的能带下移,最终形成内建电场(从ZnWO₄指向BiOBr)。这一过程直接反映在功函数变化上:BiOBr因接收电子导致其表面电子密度增加,功函数降低;而ZnWO₄因失去电子,功函数升高。
这种电荷重新分布促使光生电子(e⁻)向ZnWO₄的CB迁移,空穴(h⁺)向BiOBr的VB聚集,从而有效抑制了e⁻-h⁺复合(与PL光谱中异质结荧光强度降低的实验结果一致)。此外,能带弯曲形成的势垒进一步加速了载流子的空间分离,解释了该异质结光催化效率提升的机理。
HOMO – LUMO
最高占据分子轨道(Highest Occupied Molecular Orbital,HOMO)和最低未占据分子轨道(Lowest Unoccupied Molecular Orbital,LUMO)是分子轨道理论中的重要概念。
HOMO 是分子中能量最高的被电子占据的分子轨道,LUMO 是能量最低的未被电子占据的分子轨道,它们之间的能量差称为能隙(HOMO – LUMO Gap) 。在 DFT 计算中,可以通过求解 Kohn – Sham 方程得到分子的分子轨道能级,从而确定 HOMO 和 LUMO 。
为什么可以分析电荷转移
在化学反应和光物理过程中,电子很容易从 HOMO 跃迁到 LUMO 。当分子与其他物质发生相互作用时,如果存在合适的能量条件,电子会从 HOMO 转移到其他物质的空轨道上,或者从其他物质的占据轨道上获得电子填充到 LUMO 中,从而发生电荷转移 。
HOMO – LUMO 能隙的大小反映了分子得失电子的难易程度,能隙越小,分子越容易发生电荷转移 。例如,在有机光伏材料中,给体材料的 HOMO 能级与受体材料的 LUMO 能级之间的能量匹配对于电荷分离和转移至关重要 。
应用
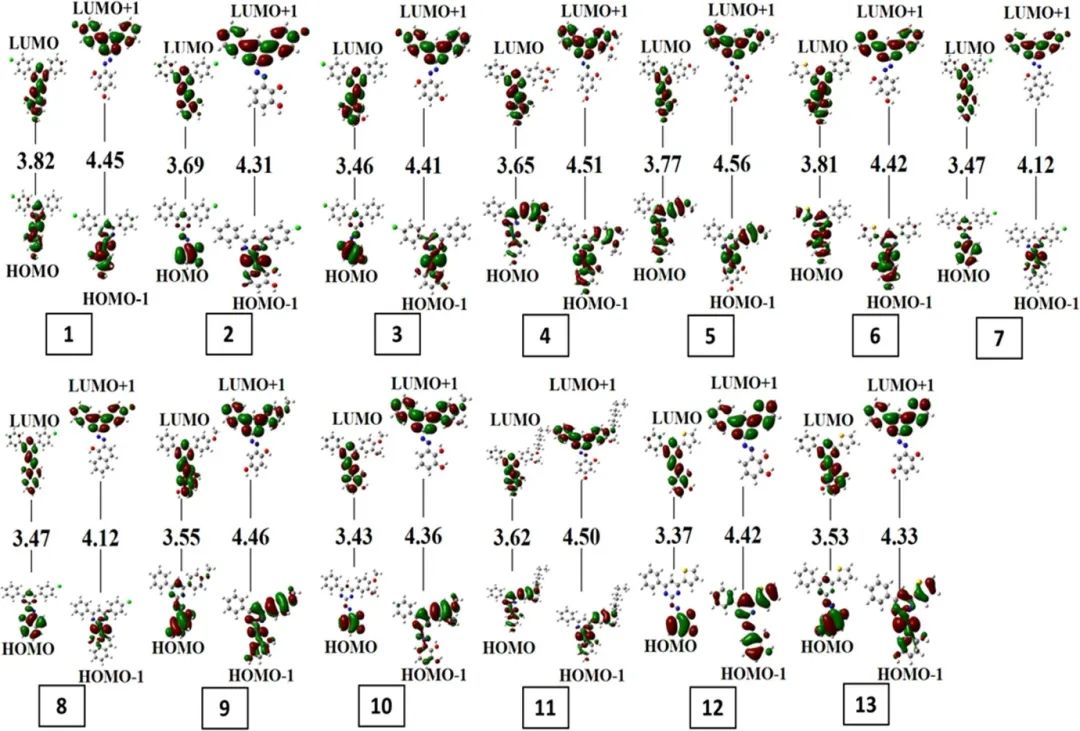
通过分析HOMO(最高占据分子轨道)和LUMO(最低未占分子轨道)能级,揭示了分子体系中的电荷转移机制。HOMO-LUMO能隙的大小直接反映了电子转移的难易程度:较小的能隙(如分子1的2.57 eV)表明电子更容易从HOMO跃迁到LUMO,从而促进电荷转移过程。具体来看,分子1-5与6-eolinks之间的能隙差异(如分子1的较低能隙)说明这些分子更倾向于作为电子给体,将电子转移到能级匹配的受体分子上。
此外,HOMO能级的位置(如Macauromondo的-9.97 eV)和LUMO能级(如-0.03 eV)的相对能量差决定了电荷转移的方向性——电子通常从高能级的HOMO流向低能级的LUMO。这种能级匹配关系在远程桥接的2,3,8-核体系中尤为明显,进一步证实了HOMO-LUMO分析在预测电荷转移路径中的重要性。
因此,通过HOMO-LUMO能级和能隙的分析,可以清晰地揭示分子内或分子间电荷转移的驱动力和方向性,为理解电子转移过程提供了关键的理论依据。
静电势
静电势(Electrostatic Potential)是指在空间某一点处,将一个单位正电荷从无穷远处移动到该点时所做的功,它是描述体系电子结构的一个重要物理量 。在 DFT 计算中,静电势可以通过求解泊松方程从电子密度计算得到,其数学表达式为:
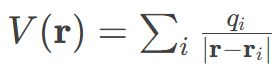
其中,V(r) 表示空间r处的静电势,其中 qi 为体系中各原子或电荷的贡献 。静电势反映了体系中电子和原子核的电荷分布对空间电场的影响 。
为什么可以分析电荷转移
静电势能够分析电荷转移,关键在于其直观体现电子给/受能力。当两个组分(如分子与基底)接近时,电荷倾向于从高静电势区(电子供体)流向低静电势区(电子受体)。
例如,若金属原子的ESP显著高于gC₃N₄的氮位点,电子将从金属向基底转移,形成离子键;反之则可能导致反向转移。此外,ESP的梯度(如静电势差ΔV)可量化电荷转移驱动力,而静电势面的极小值点(如分子的负电区域)常预示亲核反应位点。
在界面研究中,ESP分布与差分电荷密度相互印证:若某区域ESP下降伴随电荷积累(差分图中黄色),则证实电子注入;反之则对应电子流失。因此,结合ESP与电荷密度差分,可全面揭示电荷转移的方向、强度及成键本质(如共价、离子或混合键合),为催化、吸附等过程提供关键电子结构信息。
应用
分子静电势分析揭示了偶氮染料分子中电荷转移的驱动力和反应位点倾向。从图中的颜色分布来看,蓝色区域(高正电势)主要集中于氢原子周围,表明这些位点因核电荷暴露而呈现缺电子特性,易受亲核攻击并作为电子受体;而红色区域(高负电势)集中在N、O、S和Cl等电负性原子上,显示其富电子特性,可作为电子供体或亲电位点。
例如,偶氮基团(—N=N—)附近的负电势区暗示其可能通过孤对电子参与电荷转移,而苯环上的正电势氢则可能通过静电吸引接受电子。MEP的数值梯度(如+7.375×10⁻²至−7.832×10⁻²)进一步量化了电荷转移的不对称性:电势差越大,电子从负电势区(如硫原子)向正电势区(如质子化位点)转移的趋势越强。
这种分析不仅预测了分子间相互作用(如氢键或亲电/亲核反应)的活性位点,还解释了偶氮染料在光催化或降解过程中可能的电荷分离路径——电子从富电子发色团(如偶氮基)流向缺电子受体(如吸附的氧化剂),为设计高效降解策略提供了电子尺度依据。
态密度
态密度(Density of States, DOS) 是描述材料中电子能级分布的函数,表示单位能量区间内允许的电子量子态数量。态密度可分为总态密度(TDOS)和分波态密度(PDOS),后者进一步分解为原子轨道(s、p、d、f)的贡献,从而揭示不同轨道的电子结构特征。
为什么可以分析电荷转移
当两个体系(如金属原子与基底)相互作用时,其电子结构会发生重构,导致DOS峰位移动、新峰出现或峰形变化。
例如,若掺杂原子的d轨道峰向费米能级(EF)下方移动,表明电子从该原子流向基底(电荷耗散);反之,若峰向 EF 上方移动,则可能发生反向转移(电荷积累)。此外,杂化峰(如金属d-N p的耦合峰)的出现直接反映化学键形成,其强度与电荷共享程度相关。
通过对比孤立组分与复合体系的DOS,可定量评估电荷转移量及成键性质(共价、离子或混合键),从而揭示界面相互作用机制。
应用
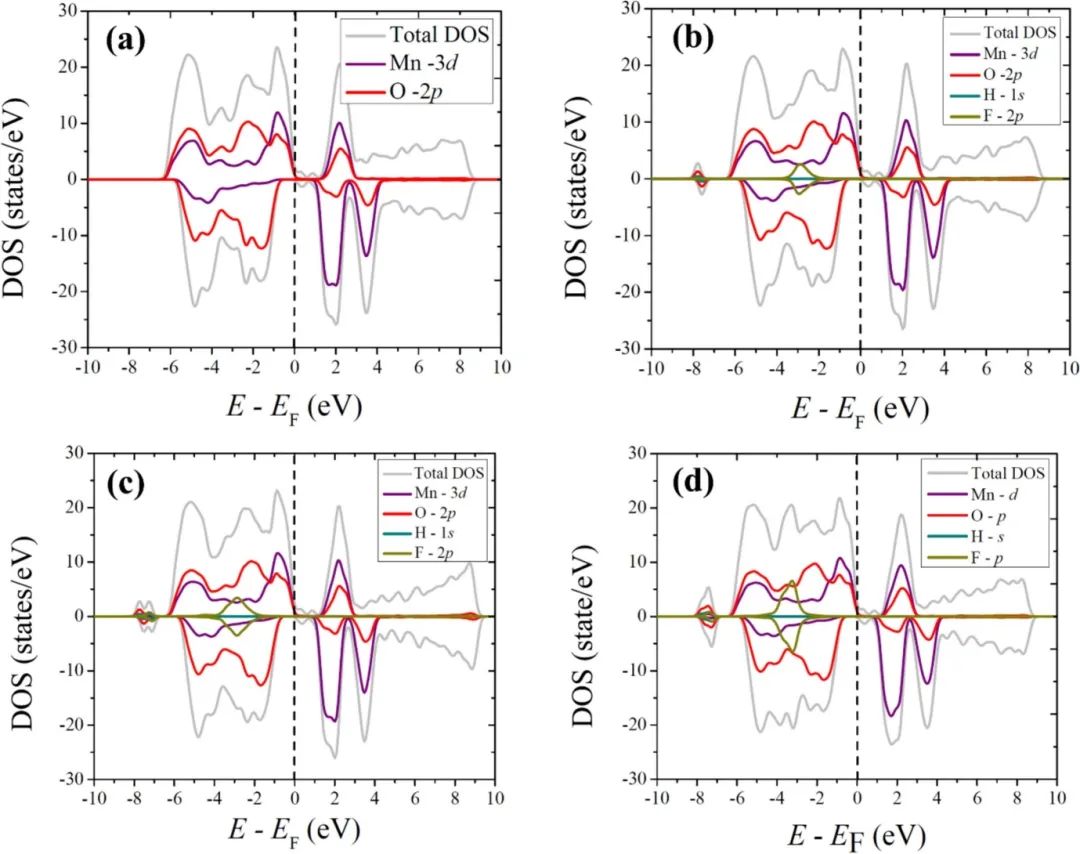
通过投影态密度(PDOS)分析揭示了HF分子在Li₂MnO₃(001)表面吸附时的电荷转移机制。PDOS结果显示,Mn的d轨道在费米能级附近的贡献几乎未受HF吸附影响(图a-c),表明Mn未直接参与电荷交换,与Bader电荷分析中Mn仅+0.02e的微小变化一致。
关键变化集中在F和H的轨道上:F原子在-7.5 eV附近出现新峰(图b),对应其p轨道与表面Li的杂化,证实了F-Li的离子键相互作用;而H原子在-7 eV附近的峰(图c)则指向H-O键的形成,解释了实验中H从HF解离后与表面O的键合。
值得注意的是,随着HF覆盖度增加,F相关峰强度增强(0.06 eV处),表明更高覆盖度下F的电子局域化更显著,可能增强表面钝化效应。此外,费米能级附近未出现金属性新态,说明HF吸附未引入额外导电通道,而是通过F和H的轨道重构实现电荷再分配,降低了材料反应活性。
态密度分析因此清晰地揭示了电荷转移的原子尺度路径:电子从Li流向F形成极性键,同时H与O键合,而Mn保持惰性,这一机制为理解表界面修饰对材料电子结构的调控提供了重要依据。
综上所述,差分电荷密度、Bader 电荷等电荷布局、功函数、HOMO – LUMO、静电势和态密度这六种方法从不同角度为 DFT 计算中分析电荷转移提供了有力的工具。它们各自具有独特的定义和分析原理,在材料科学、化学、物理学等领域有着广泛的应用。
通过综合运用这些方法,可以更全面、深入地理解电荷转移过程,为新材料的设计和性能优化提供坚实的理论基础。
在未来的研究中,随着 DFT 计算方法的不断发展和完善,这些分析方法也将不断拓展其应用范围和深度,为科学研究和技术创新提供更强大的支持 。