

引言
在钠离子电池的研究中,正极材料对电池的性能起着决定性作用。层状正极材料因其独特的结构和优异的电化学性能,成为钠离子电池正极材料研究的重点。
这类材料具有较高的理论容量和能量密度,能够为钠离子电池提供良好的储能基础。此外,层状结构所具备的二维钠离子扩散通道,有助于实现较快的离子扩散动力学,提升电池的充放电速率。
理论研究在材料开发过程中具有不可替代的作用。密度泛函理论(DFT)计算作为一种强大的理论研究工具,能够从原子和电子层面深入理解材料的本征性质,为实验设计提供理论指导,加速新型材料的研发进程。
在钠离子电池层状正极材料的研究中,DFT计算能够揭示材料的钠离子扩散能垒、电子结构、相稳定性等关键性质,为优化材料性能、开发新型层状正极材料提供重要依据。
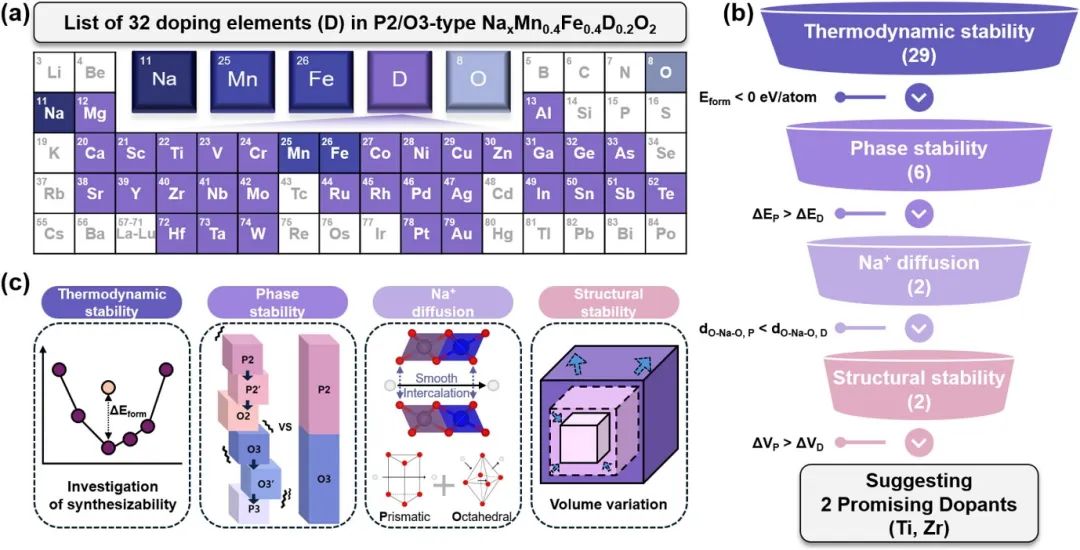

钠离子电池层状正极材料的定义
基本概念
钠离子电池层状正极材料的化学通式通常表示为 NaxMO₂,其中 M 代表过渡金属元素,如 Co、Fe、Mn、Ni 等 。这类材料具有典型的层状结构,根据其晶体结构中钠离子(Na⁺)的排列方式和配位环境的不同,常见的层状材料可分为 O3 型和 P2 型。
O3 型层状材料中,钠离子位于过渡金属氧八面体(MO6)层之间的八面体(Octahedral)间隙位置,“O” 即代表八面体配位。例如 NaCoO₂,其结构中钠离子在层间以八面体配位方式排列,具有较高的对称性和结构稳定性。
P2 型层状材料中,钠离子位于过渡金属氧三棱柱(MO6)层之间的三棱柱(Prismatic)间隙位置,“P” 代表三棱柱配位。以 Na₂/3Fe₁/2Mn₁/2O₂为例,其结构中钠离子在层间以三棱柱配位形式存在,这种结构使得 P2 型材料在钠离子嵌入和脱出过程中表现出独特的电化学行为 。
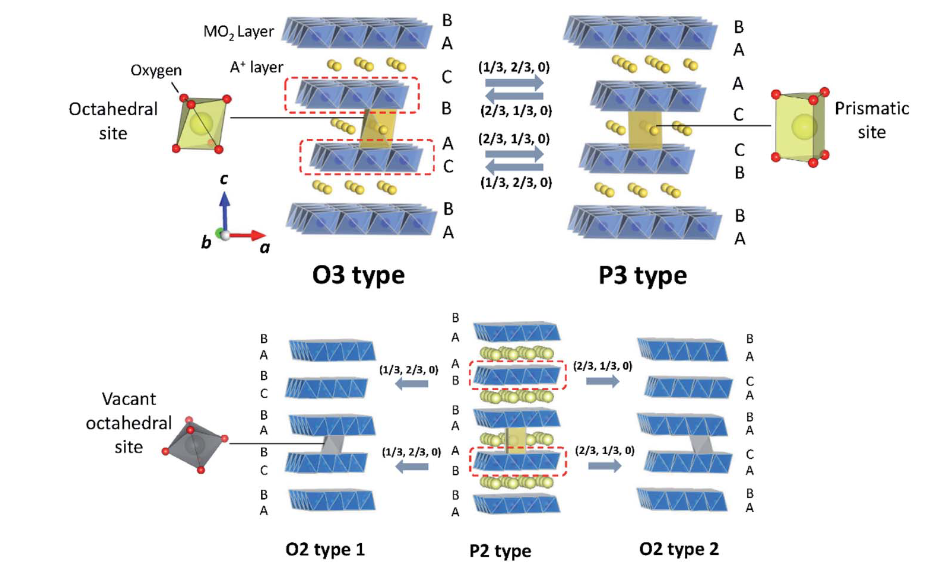
结构特征
钠离子电池层状正极材料的层状结构具有明显的特征,其过渡金属层与钠层交替排列,形成了二维钠离子扩散通道。在这种结构中,过渡金属离子被氧离子紧密包围,构成过渡金属氧层,这些过渡金属氧层通过共享氧原子相互连接,形成稳定的骨架结构。而钠离子则分布在过渡金属氧层之间的间隙位置,形成钠层。
这种二维钠离子扩散通道为钠离子的迁移提供了便捷的路径。当电池充放电时,钠离子能够在层间的扩散通道中快速嵌入和脱出,实现电池的电化学功能。同时,过渡金属层与钠层的交替排列也保证了材料结构的稳定性,使得层状正极材料在多次充放电循环过程中能够保持相对稳定的晶体结构,从而为电池的长循环寿命提供保障。
层状正极材料的特点
优势
1. 高理论容量和能量密度
钠离子电池层状正极材料具有较高的理论容量和能量密度。以常见的 O3 型和 P2 型层状材料为例,在钠离子完全脱出的情况下,它们能够提供较高的比容量。这是因为层状结构中大量的钠离子可以参与氧化还原反应,实现电荷的存储和释放。
较高的比容量意味着在相同质量或体积的正极材料中,能够存储更多的电能,进而提高电池的能量密度,满足不同应用场景对电池高能量密度的需求。
2. 较快的 Na⁺扩散动力学
二维钠离子扩散通道赋予了层状正极材料较快的 Na⁺扩散动力学。钠离子在层间的扩散路径相对较短且较为顺畅,扩散阻力较小。这使得在电池充放电过程中,钠离子能够快速地在正极材料中嵌入和脱出,从而提高电池的充放电速率。
与一些具有三维复杂扩散通道的材料相比,层状正极材料在高倍率充放电条件下能够更好地保持电化学性能,满足快速充电等应用场景的需求。
3. 稳定的晶体结构
层状正极材料的晶体结构具有一定的稳定性。其过渡金属层与钠层交替排列的结构特点,使得在钠离子嵌入和脱出过程中,能够在一定程度上保持晶体结构的完整性。
尽管在深度脱钠过程中,材料可能会发生相变,但相较于其他类型的正极材料,层状结构的材料在结构稳定性方面仍具有一定优势。稳定的晶体结构有助于提高电池的循环性能,减少因结构坍塌或相变导致的容量衰减,延长电池的使用寿命 。
DFT 计算的具体应用案例
钠离子迁移路径与扩散机制
在研究钠离子在层状正极材料中的迁移路径与扩散机制时,DFT 结合 NEB 方法是常用的手段。以 O3 型 NaCoO₂材料为例,通过构建包含多个晶胞的超胞模型,利用 NEB 方法计算钠离子在层间的扩散路径 。
计算结果表明,钠离子在 O3 型 NaCoO₂中沿着特定的八面体间隙位置迁移,其扩散能垒约为 0.3 eV 。这一结果表明钠离子在该材料中的扩散相对较为容易,与实验中观察到的较好的充放电性能相吻合。
进一步研究发现,当对 NaCoO₂材料进行掺杂改性时,如掺杂少量的 Mn 元素,钠离子的扩散路径和扩散能垒会发生变化。
DFT 计算显示,Mn 掺杂后,钠离子的扩散能垒降低至约 0.25 eV,这是由于 Mn 掺杂改变了材料的局部结构和电子结构,使得钠离子迁移的能量势垒降低 。这些计算结果为解释掺杂对材料离子扩散性能的影响提供了理论依据,也为通过掺杂优化材料性能提供了指导。
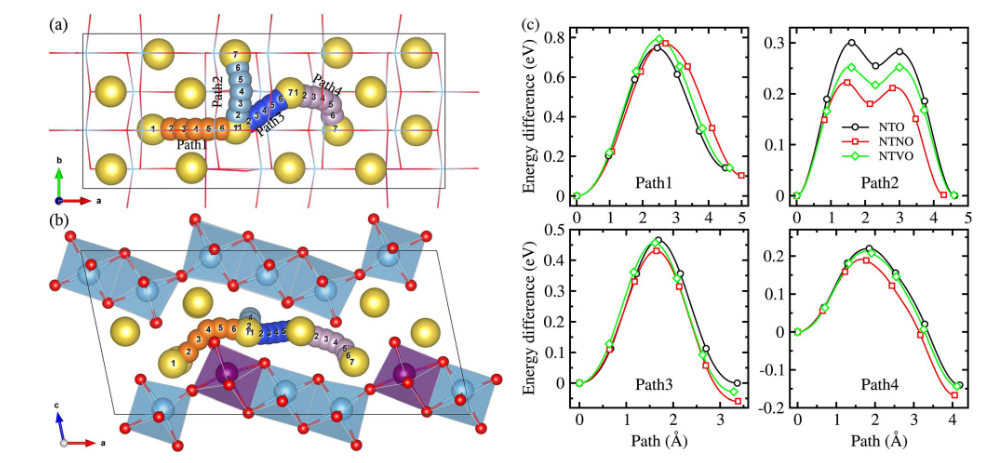
材料稳定性分析
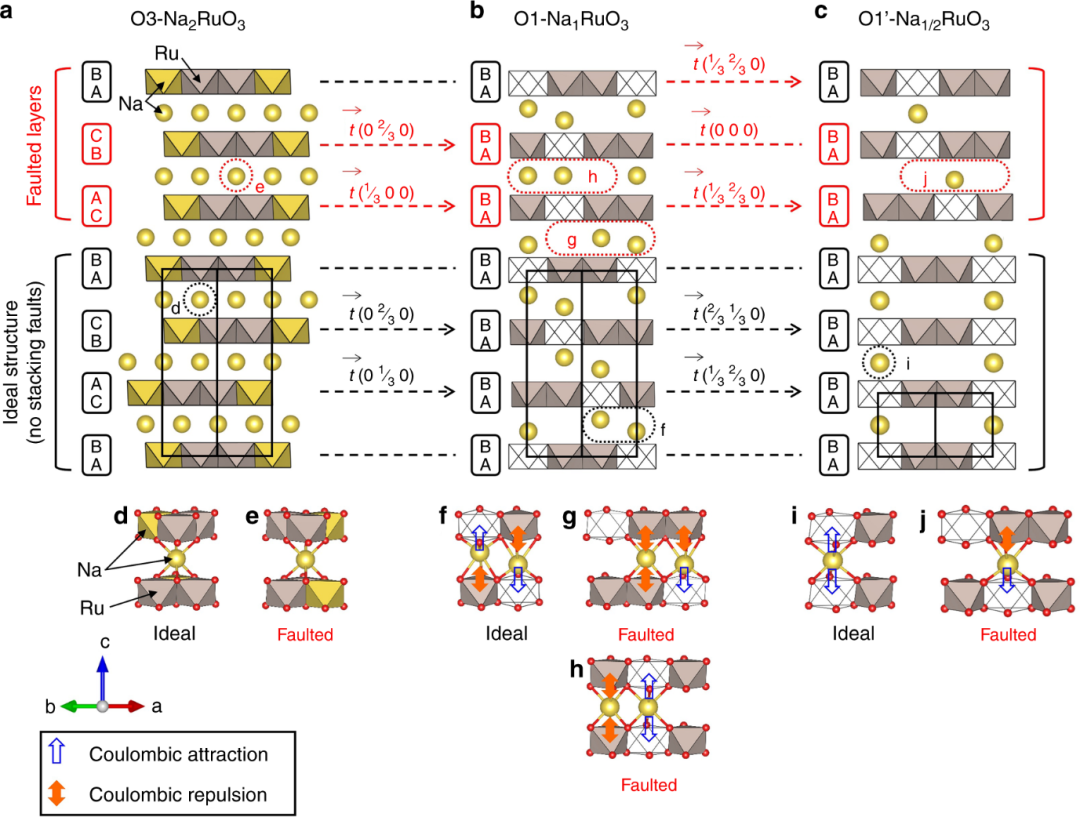
通过对比Na₂RuO₃在充电过程中不同钠含量(x=2、1、1/2)下的结构演变,揭示了层状正极材料中独特的“自修复”机制及其稳定性根源。
研究发现,随着钠离子的脱出(充电),材料从初始的O3-Na₂RuO₃(40%堆垛缺陷)依次转变为O1-NaRuO₃(10%缺陷)和O1′-Na₁/₂RuO₃(仅2%缺陷),堆垛缺陷率显著降低。
这一现象源于三维库仑相互作用驱动的结构自有序化:在脱钠过程中,[Ru₂/₃□₁/₃]O₂层(□为空位)与相邻钠层的Na⁺-□(空位)库仑吸引力增强,同时Na⁺-Ru⁵⁺库仑排斥力迫使钠离子向空位偏移,协同作用促使层间滑移(O3→O1→O1’相变)以优化静电平衡,从而修复初始堆垛缺陷。
此外,O1和O1’相中Na⁺的局部配位环境(如NaO₆八面体与空位或RuO₆的共享面)进一步抑制了缺陷形成,而完全脱钠的O1′-Na₁/₂RuO₃因高度有序的[Na₁/₃□₂/₃]层与[Ru₂/₃□₁/₃]O₂层交替堆叠,实现了近乎无缺陷的结构。
这种“充电诱导有序化”机制不仅提升了材料的结构可逆性,还通过空位有序化稳定了氧氧化还原反应,为设计高容量、长寿命的钠离子电池正极材料提供了新思路——即通过调控碱金属空位的有序性和层间库仑作用,实现结构稳定与电化学性能的协同优化。
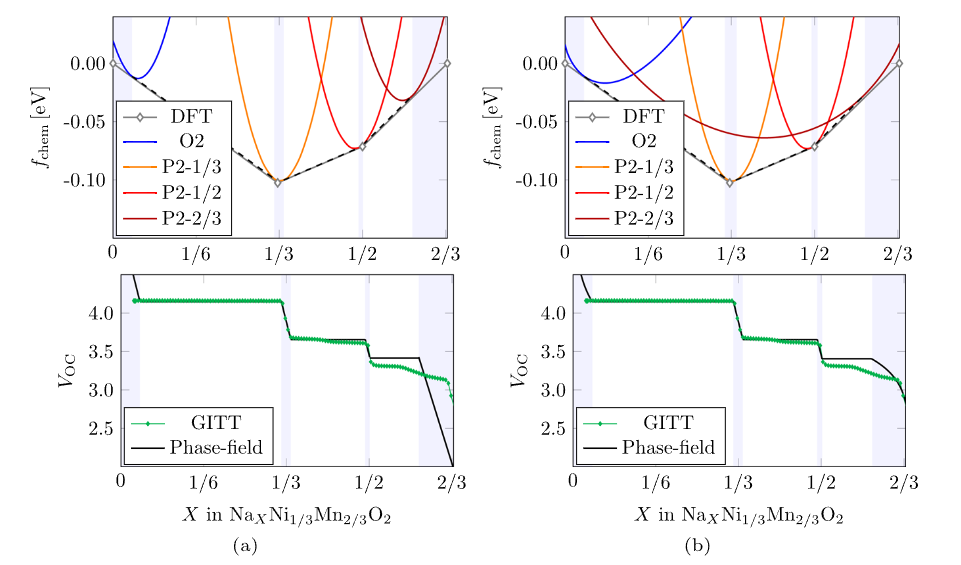
电压与容量预测
DFT计算可以准确预测层状正极材料的电压与容量。通过计算钠离子嵌入脱出反应的吉布斯自由能变化,结合能斯特方程,可以得到材料的平均电压 。
以 O3 型 NaNi₁/3Mn2/3O₂材料为例,DFT计算了钠离子从材料中逐步脱出过程中的能量变化,进而计算出材料的电压曲线 。计算得到的电压曲线与实验测量结果具有较好的一致性,表明 DFT 计算在预测材料电压性能方面的有效性。
在容量预测方面,DFT计算可以确定材料中能够参与氧化还原反应的钠离子数量,从而预测材料的理论容量 。通过分析材料的电子结构和氧化还原反应机理,计算出不同过渡金属离子在反应过程中的电子转移数,结合材料的化学组成,得到材料的理论比容量。这些理论预测结果为评估材料的储能性能提供了重要参考,有助于筛选出具有高电压和高容量潜力的层状正极材料。
掺杂改性设计
DFT计算在层状正极材料的掺杂改性设计中发挥了重要作用。为了提高材料的电化学性能,研究人员尝试通过掺杂不同的过渡金属离子进行改性 ,DFT计算了 Ni、Co 等过渡金属离子掺杂对材料性能的影响。
计算结果表明,Ni 掺杂可以降低材料的钠离子扩散能垒,提高材料的电子导电性,同时增强材料的结构稳定性 。这是因为 Ni 离子的掺杂改变了材料中过渡金属层的电子结构和离子间相互作用,优化了钠离子的扩散环境和材料的整体性能。
基于 DFT计算结果,实验研究人员进行了相应的掺杂实验,成功制备出了 Ni 掺杂的 P2 型 Na₂/3Fe₁/2Mn₁/2O₂材料,并验证了其在电池中具有更好的电化学性能,如更高的比容量和更长的循环寿命 。
这一案例充分展示了 DFT 计算在指导层状正极材料掺杂改性设计方面的重要意义,通过理论与实验的结合,能够加速高性能钠离子电池层状正极材料的开发。