

密度泛函理论(DFT)通过键能(断裂所需能量)、键长(几何优化测定)、键级(NBO分析量化成键电子数)评估化学键稳定性。
结合电子结构工具如COHP(能量贡献分析)、Bader电荷(原子电荷量化)、AIM理论(键临界点性质)及HOMO-LUMO能隙(分子稳定性),系统解析成键本质。
经典案例中,本征键强度指数(IBSI)耦合键解离能与键级,预测高能化合物热分解误差仅2.3%。机器学习与多尺度模拟进一步推动动态环境下的精准评估。


化学键稳定性核心参数

键能(Bond Energy)
键能作为化学键稳定性的核心参数,通过密度泛函理论(DFT)计算可精准量化断裂特定化学键所需的能量。
计算流程包括几何优化分子基态结构,计算其总能量(E_molecule),解离为孤立气态原子后求总能量(E_atoms),键能即为两者差值(BE = E_atoms – E_molecule),并结合零点能校正(ZPE)消除量子振动基态能级的影响。
例如,H₂O中O-H键能计算需优化H₂O结构,解离为2H与1O,经ZPE校正后与实验值高度吻合。
DFT计算的键能数据不仅揭示分子热力学稳定性,还可预测反应活性方向(如低键能优先断裂),为催化剂设计、材料降解机制分析及反应路径优化提供定量理论依据。
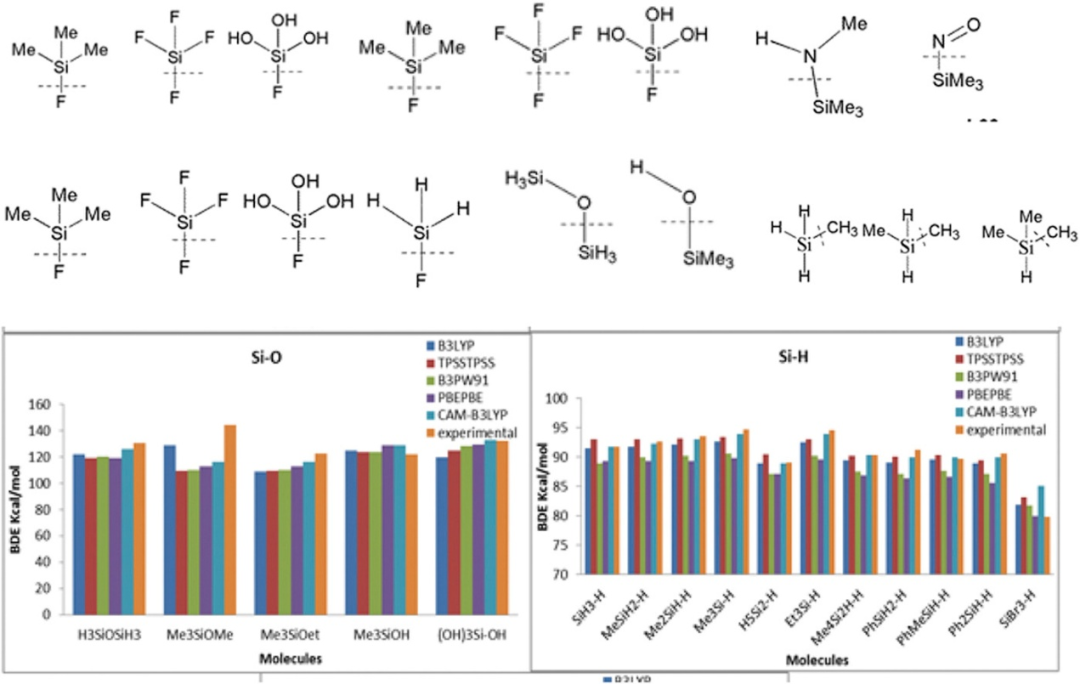
DOI:10.1016/j.molstruc.2017.04.066
键长(Bond Length)
键长作为化学键稳定性的重要指标,通过密度泛函理论(DFT)的几何优化模块(如B3LYP泛函与6-311++G(d,p)基组)可精确测定,其与键能呈负相关——键长缩短通常伴随键能增加。
例如,碳-碳单键(C-C)优化键长约为1.54 Å,对应键能346 kJ/mol,而三键(C≡C)键长缩短至1.20 Å,键能显著提升至839 kJ/mol,揭示键级升高对稳定性的强化作用。
DFT计算通过求解电子基态波函数确定原子核平衡位置,结合振动频率分析验证势能面极小点,确保键长数据的可靠性(误差)。
在材料设计中,短键长特征赋予高机械强度与热稳定性,而长键长可能预示易断裂倾向,指导功能材料的理性修饰(如掺杂或应变调控)。
键长分析还可关联电子结构特性,如金属-有机框架中配位键长变化反映配位强度差异,进而影响催化活性与载流子迁移率。这类计算策略为化学键稳定性评估与材料性能优化提供了原子尺度的定量依据。
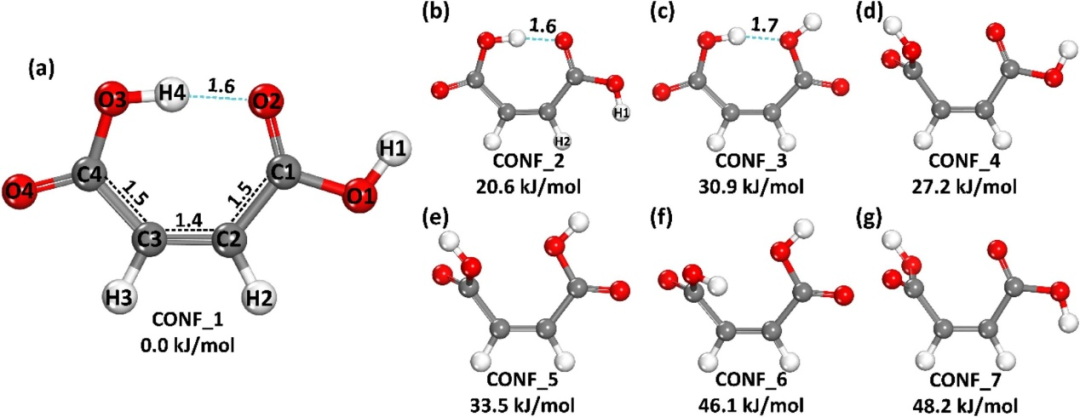
DOI:10.1016/j.jcat.2025.116212
键级(Bond Order)
键级(Bond Order)通过量化化学键的成键电子数目,直接反映键的强度与稳定性,是解析分子或材料化学性质的核心参数。
基于密度泛函理论(DFT)的自然键轨道(NBO)分析可精确计算键级:通过分解分子轨道为定域化的自然键轨道,统计成键与反键轨道的电子占据数差,例如乙烷(C-C单键)的键级为1,对应电子占据差1.98,而乙烯(C=C双键)与乙炔(C≡C三键)的键级分别增至2与3,对应键能显著提升(从346 kJ/mol至839 kJ/mol)。
在过渡金属配合物中,NBO分析可解析金属-配体键的键级变化(如Fe-CO键级从1.2至1.8),揭示配体场强度对催化活性的调控机制;在材料科学中,石墨烯的sp²杂化C-C键级为1.5(介于单双键之间),赋予其高导电性与力学稳定性。
通过键级计算,可预测反应选择性(如低键级位点优先断裂)、指导催化剂设计(如优化活性中心键级以增强中间体吸附),并为复杂体系(如自由基或激发态)的电子结构分析提供定量依据,成为连接理论计算与实验观测的关键桥梁。
DOI:10.1007/s00894-024-05877-5
从电子结构到键稳定性
COHP分析
COHP(晶体轨道哈密顿布居分析)作为基于密度泛函理论(DFT)的化学键能量解析工具,如同一位微观世界的 “能量会计师”,通过量化原子间轨道相互作用的能量贡献,精准建立起化学键成键强度与稳定性的直接关联。
其核心价值在于打破传统态密度(DOS)仅能描绘电子分布 “地图” 却无法解读键合 “密码” 的局限,将电子结构的能量信息与化学键的本质特征编织成一张逻辑严密的分析网络 —— 既能从轨道作用的能量起伏中辨别成键与反键的竞争态势,也能在能级交叠的细微变化里捕捉键稳定性的深层动因。
这种“从能量流动看键合本质” 的独特视角,让研究者得以穿透电子云的迷雾,在原子间作用力的微观剧场中,观察轨道相互作用如何通过能量的分配与传递,书写化学键的强弱兴衰史,为材料电子结构解析与成键机制研究提供了一把兼具深度与精度的 “能量标尺”。
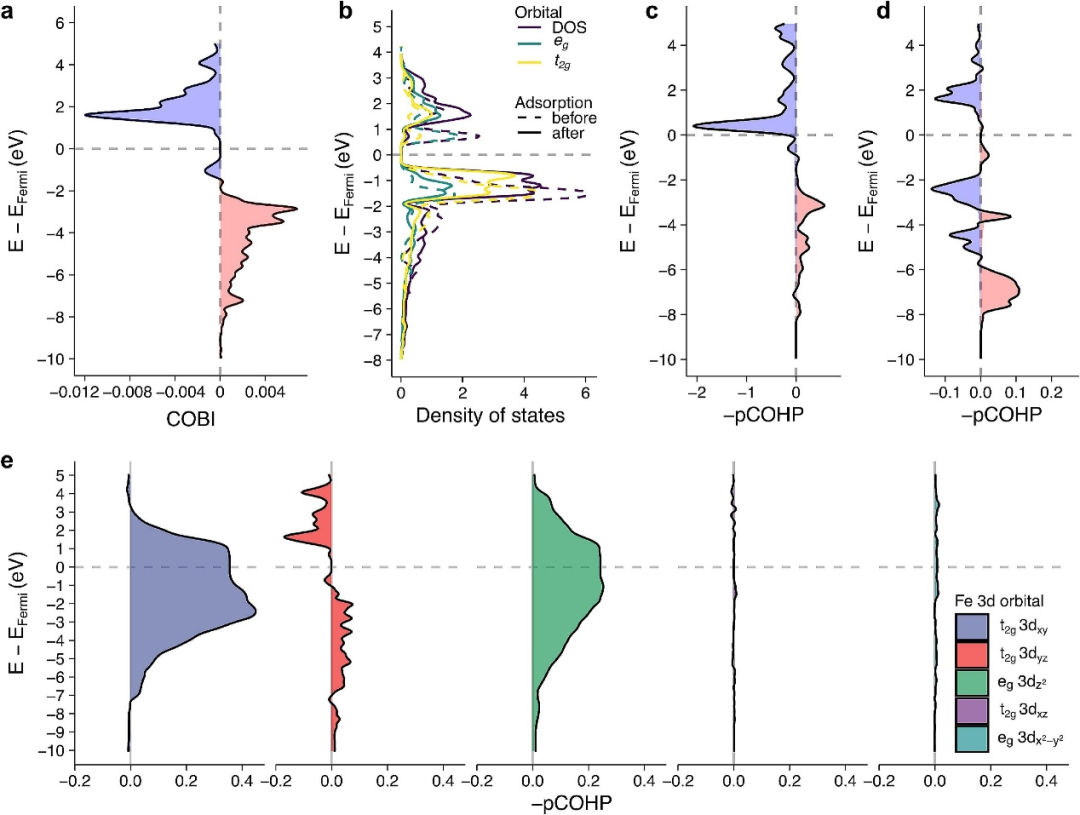
DOI:10.1016/j.apsusc.2025.163594
自然键轨道(NBO)分析
自然键轨道(NBO)分析如同分子内的电荷侦探,专注于揭示原子间电荷转移的秘密与成键特征的微观剧本。
它借助第二阶微扰理论构建起一套精密的“能量账本”,通过计算稳定化能量(E (2))定量刻画供体 – 受体轨道间的相互作用强度 —— 这一过程如同在分子轨道的 “对话” 中测量能量的传递效率:当 σ 轨道向 σ* 反键轨道发生电子离域时,E (2) 数值越大,意味着轨道间的 “协同效应” 越强,化学键的稳定性便如同被注入更多 “能量锚点”。
例如,当表格中出现 E (2)=5.00 kcal/mol 的标注时,不仅代表着供体 – 受体轨道间存在显著的稳定化作用,更暗示着分子通过轨道相互作用实现了能量优化的 “智慧选择”。
这种从电荷转移到轨道相互作用的量化分析,让研究者能够在分子的电子网络中追踪能量流动的轨迹,如同破译一部用轨道符号书写的“成键密码”,为理解分子构象稳定性、反应活性等核心化学问题提供兼具直观性与深度的分析工具。
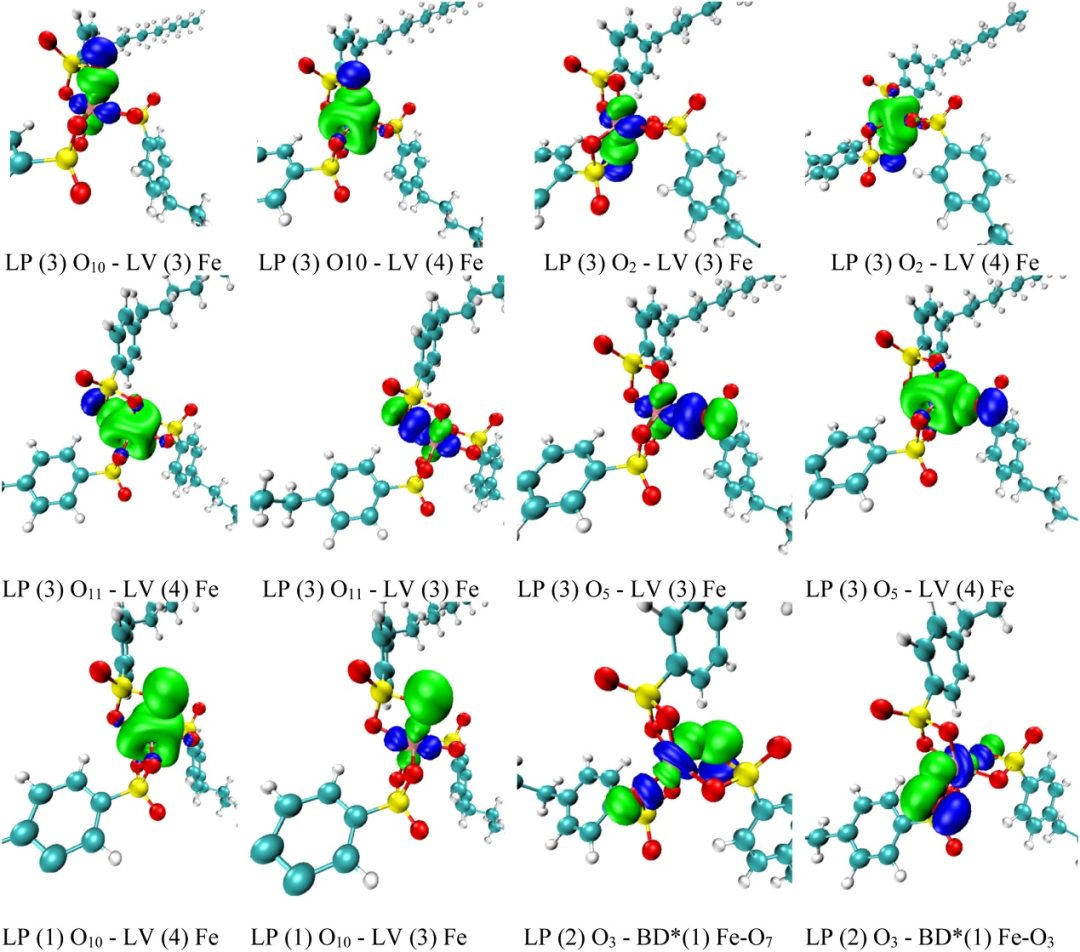
DOI:10.1016/j.cdc.2025.101186
Bader电荷分析
Bader 电荷分析如同一位原子级的“电子密度测绘师”,基于量子化学中电子密度的拓扑学划分,为分子内各原子区域构建起精确的电荷“地籍图”。
该方法通过识别电子密度梯度的极值点与零通量面,将分子空间划分为独立的原子盆地,进而量化每个原子的电荷分布—— 这种 “以电子密度轮廓定界” 的分析方式,如同在分子的电子云海洋中勾勒出原子疆域的海岸线。
例如在典型离子键体系中,钠原子(Na⁺)的电荷值接近 + 0.9 e,氯原子(Cl⁻)约为 – 0.9 e,清晰展现出电荷的定向转移;而在共价键场景下,碳原子的电荷通常仅为 ±0.2 e,揭示出成键电子的均衡共享特征。
这些数值不仅是原子得 / 失电子能力的量化体现,更能为判断化学键的离子性 – 共价性连续谱提供直接依据。
从固态材料的极性分析到化学反应中的电荷迁移追踪,Bader 电荷分析以电子密度拓扑为 “标尺”,让分子内的电荷分布从抽象的理论描述转化为可解读的数字语言,为理解成键本质、预测分子性质搭建起从微观电子结构到宏观化学行为的桥梁。
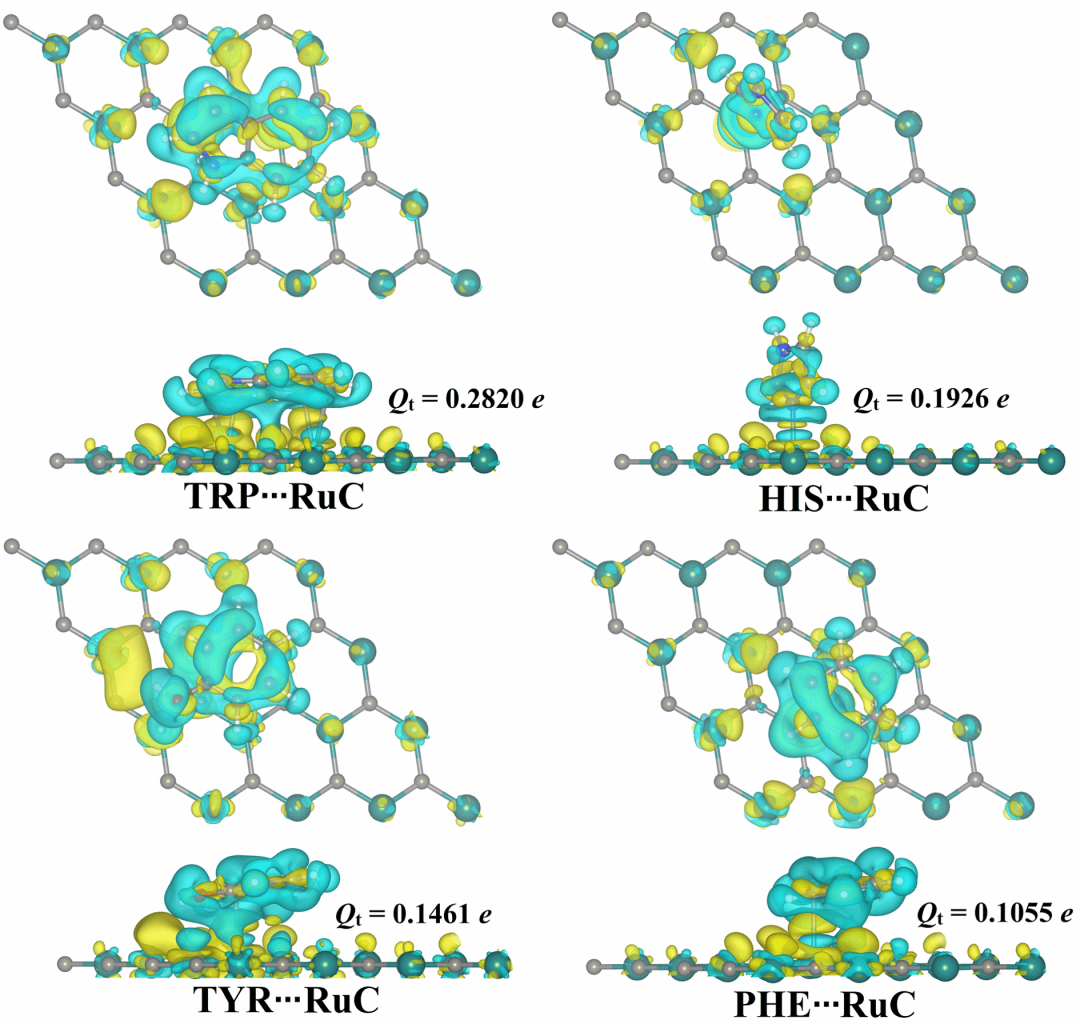
DOI:10.1039/d4na00670d
分子轨道能量与HOMO-LUMO能隙
HOMO(最高占据分子轨道)与LUMO(最低未占分子轨道)的能隙(ΔE=ELUMO−EHOMO)是衡量分子稳定性和化学反应活性的核心参数,能隙越大表明电子跃迁所需能量越高,分子越稳定。
例如,2,5-二甲基苯甲酸的HOMO-LUMO能隙为0.195 eV(HOMO=-5.70 eV,LUMO=-5.50 eV),显著高于苯甲酸(ΔE=0.12 eV),表明甲基取代基通过电子效应增强分子刚性,抑制电子离域化。
密度泛函理论(DFT)计算结合杂化泛函(如B3LYP)可精确预测能隙值(误差),揭示分子设计中的电子调控机制:吸电子基团(如-NO₂)通常降低HOMO能级(如硝基苯的HOMO=-8.2 eV),而供电子基团(如-OCH₃)抬高HOMO能级(-6.5 eV),从而调控光吸收阈值(如ΔE=2.5 eV对应可见光响应)。
在光催化与光电器件领域,HOMO-LUMO能隙的定向优化(如通过共轭延伸或掺杂)可调节载流子生成与传输效率,例如石墨烯量子点的窄能隙(~1.5 eV)赋予其宽谱光吸收特性,而有机半导体的能隙调控(2.0-3.0 eV)则决定其发光波长与电荷迁移率。
这类分析为分子稳定性评估、反应路径预测及功能材料设计提供了电子结构层面的定量依据。
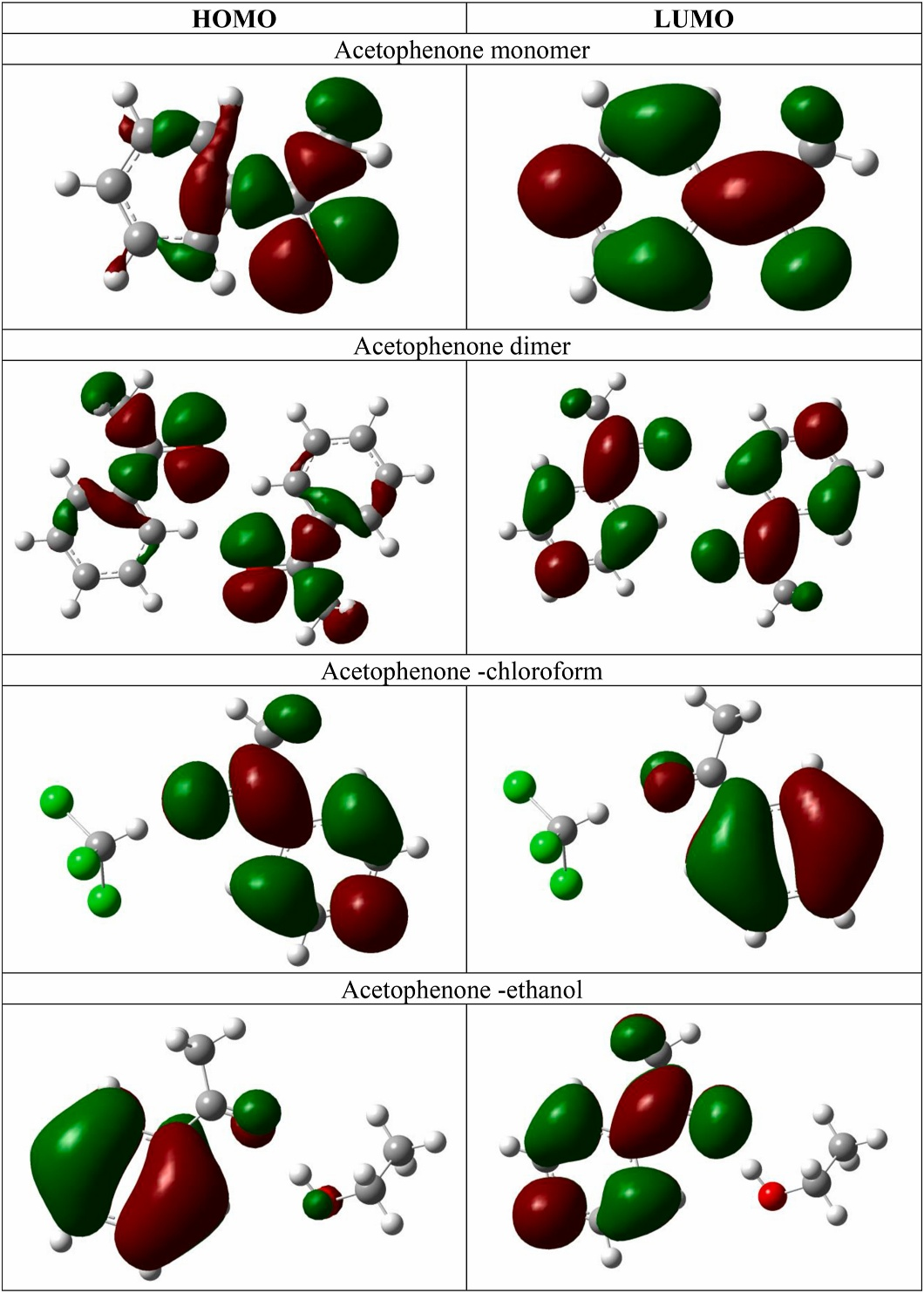
DOI:10.1016/j.optmat.2025.116683
AIM理论与键临界点分析
AIM(Atoms in Molecules)理论通过分析电子密度(ρ)及其拉普拉斯值(∇²ρ)在键临界点(BCP)的分布,定量区分化学键类型与相互作用强度。
对于共价键(如C-C),BCP处的电子密度ρ通常大于0.2 e/ų,且∇²ρ为负值(如-2.5 e/Å⁵),表明电子密度在键轴区域聚集,符合共价键的局域共享特征;氢键(如H₂O中的O-H···O)则呈现较低的ρ值(0.02-0.1 e/ų)与正∇²ρ(如+0.3 e/Å⁵),反映电子密度在氢键区域分散,验证其静电主导的弱相互作用本质。
例如,石墨烯中C-C键的ρ=0.35 e/ų、∇²ρ=-1.8 e/Å⁵,证实强共价特性;而DNA碱基对的氢键(如A-T对)ρ≈0.05 e/ų、∇²ρ=+0.2 e/Å⁵,揭示其动态可逆性。
AIM分析不仅解析传统键型(如离子键ρ=0.1-0.15 e/ų、∇²ρ>0),还可识别非经典相互作用(如卤键、金键),为分子设计(如药物靶点识别)与材料改性(如界面键合调控)提供电子密度层面的定量判据,成为连接量子化学计算与实验观测的关键桥梁。

DOI:10.1016/j.molliq.2025.127175
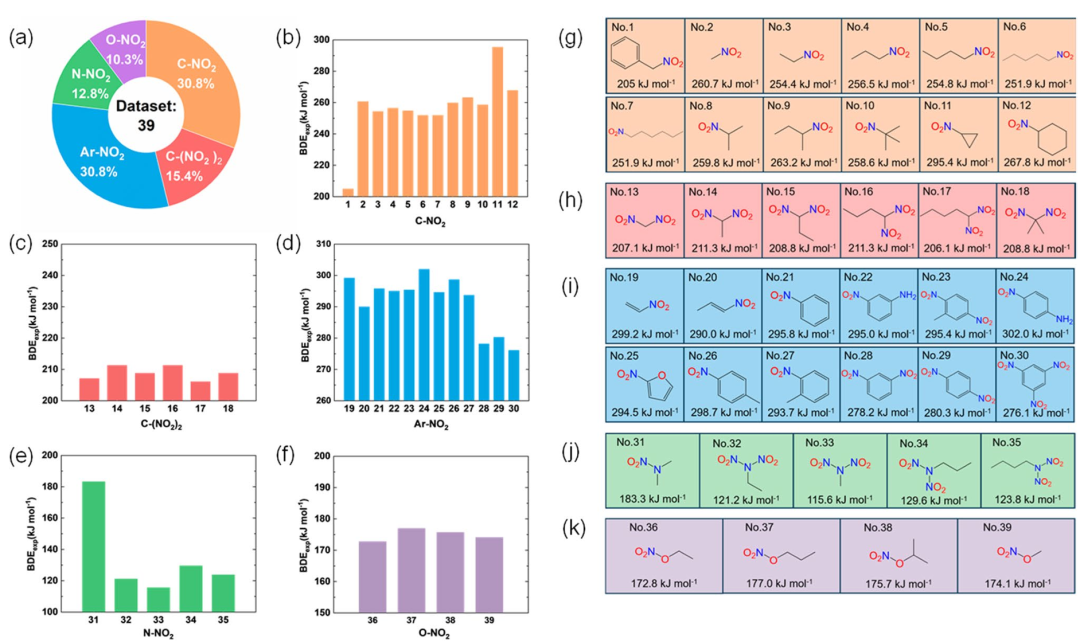
经典案例:高能化合物的键稳定性预测
在高能化合物设计领域,键稳定性预测是评估材料性能的核心挑战。Liu 等(2024)在《Journal of Molecular Modeling》中提出的本征键强度指数(IBSI)为这一问题提供了突破性解决方案。
研究团队基于密度泛函理论(DFT),通过 Gaussian 09 程序精确计算键解离能,并结合 Multiwfn 软件的键级分析功能,构建了 IBSI 这一综合评估指标。该方法通过量化键解离能与键级的耦合关系,实现了对含能化合物键稳定性的高精度预测。
研究结果显示,IBSI 与实验键解离能的相关系数 R²>0.996,表明理论预测与实验数据高度吻合。在对 24 种典型含能化合物的热分解温度预测中,IBSI 的平均误差仅为 2.3%,显著优于传统方法。
这种高精度得益于 IBSI 对化学键本质的多维度刻画:键解离能反映了断裂化学键所需的能量阈值,而键级则量化了原子间轨道重叠的程度,两者的结合使 IBSI 能够同时捕捉热力学稳定性与动力学活性。
该研究的创新之处在于将传统的键解离能计算与键级分析有机整合,形成了一套系统性的评估框架。IBSI 的提出不仅为含能材料的分子设计提供了理论指导,还为动态环境下的键稳定性研究奠定了基础。
例如,通过结合分子动力学模拟,可进一步揭示高能化合物在极端条件下的键断裂机制。这种从电子结构到宏观性质的跨尺度分析,使 DFT 计算从单纯的理论工具转变为材料研发的实用平台。
未来,随着机器学习算法的引入,IBSI 有望在保持高精度的同时大幅提升计算效率,推动高能材料领域的智能化发展。
DOI:10.1007/s00894-024-05877-5
总结
基于密度泛函理论(DFT)的计算框架,通过键能、键长、键级等基础参数的量化分析,结合COHP、自然键轨道(NBO)、Bader 电荷、原子分子静力学(AIM)等专业模块,能够系统构建化学键稳定性的评估体系。
这一体系不仅可从电子密度分布、轨道相互作用等多维度解析成键本质,更通过与现代计算技术的融合展现出广阔的发展前景:在方法优化层面,机器学习算法(如 LASSO)可将 DFT 计算量降低 90%,显著提升复杂体系的计算效率;
在模拟尺度上,DFT 与分子动力学(MD)的结合正推动动态环境下键稳定性的研究,使静态电子结构分析拓展至化学反应、材料相变等动态过程;在精度提升方面,针对金属 – 有机框架(MOF)配位键、共价有机框架(COF)骨架键等特定化学键开发的新型泛函,正不断突破传统泛函在复杂成键场景中的局限性。
这些技术进步不仅让研究者得以从量子力学层面精准解构化学键的电子结构密码,更通过理论计算与材料设计、催化反应优化的深度联动,使 DFT 超越单纯计算工具的定位,成为贯通微观电子行为与宏观物质性质的核心科学桥梁,为新能源材料开发、高效催化剂设计等前沿领域提供兼具理论深度与应用价值的解决方案。