MXene是一类由MAX相蚀刻得到的二维材料,其表面终端基团(如-F、-O、-OH、-S等)显著影响其结构和性能。
理论计算表明,卤素终端(-F、-Cl等)赋予MXene高导电性和力学稳定性,适合电池电极;氧/羟基终端(-O、-OH)可调节带隙,使其适用于半导体器件;硫族终端(-S、-Se)则因独特的吸附特性在催化领域表现突出。
终端基团通过改变键合方式、电子态密度和层间相互作用,调控MXene的电学、力学和化学性质,为其在能源存储、催化和柔性电子等领域的应用提供了多样化设计可能。
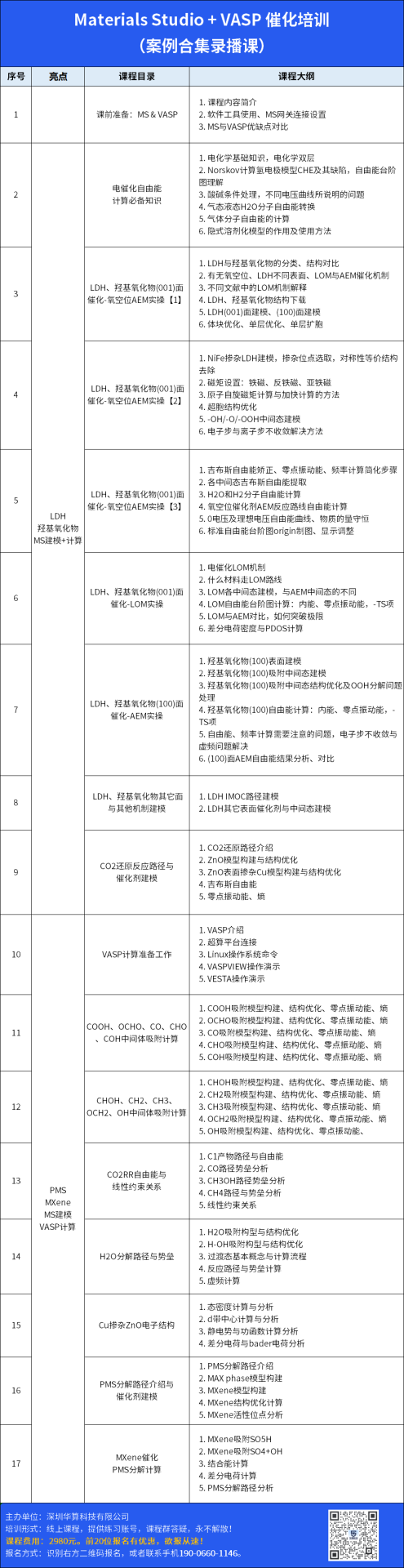
MXene是由MAX相(化学式为Mn+1AXn,其中M为过渡金属,A为Ⅲ或Ⅳ主族元素,X为碳或氮)通过选择性蚀刻A层得到的二维材料,其化学通式为Mn+1XnTx,其中Tx为表面终止基团(如-OH、-O、-F等)。
MXene的晶体结构为六方密堆积(HCP),其中过渡金属层(M)与碳/氮层(X)交替排列,表面活性位点由终止基团占据。理论计算表明,MXene的层间堆叠顺序(如ABABAB或ABCABC)及电子特性与n值和表面终止基团密切相关。
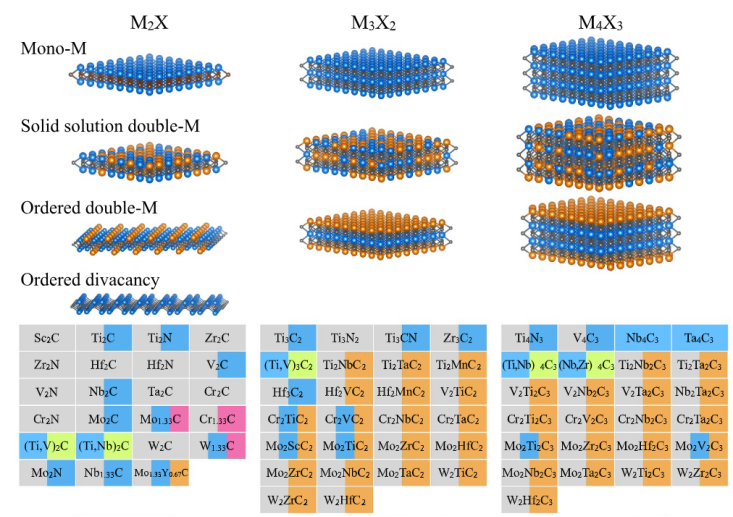

其终端原子有三种典型位点:
Top位点:位于表面M原子的顶部
Hollow C位点:位于X原子形成的六元环中心
Hollow M位点:位于M原子形成的六元环中心
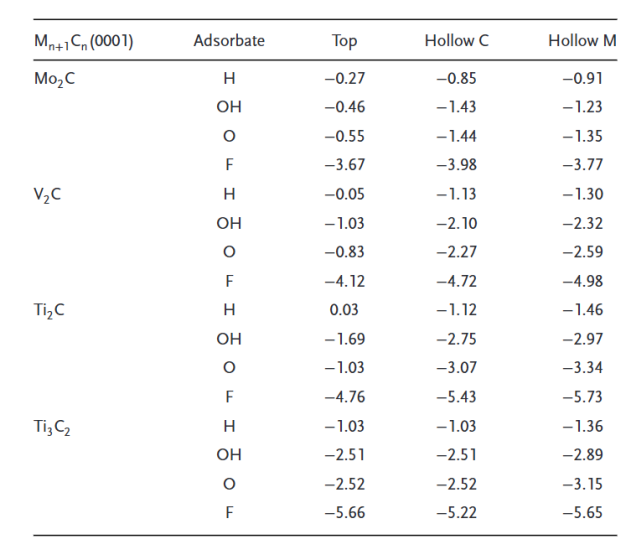
卤素终端(-F, -Cl, -Br, -I)
卤素终端(F、Cl、Br、I)对Ti₃C₂ MXene的结构、电子性质和机械性能具有显著影响,这些差异主要源于卤素的电负性、原子半径及与表面Ti的键合特性。
首先,在结构方面,随着卤素原子序数的增加(F→I),Ti–T键长和晶格常数逐渐增大,例如Ti–F键长为2.17 Å,而Ti–I键长增至2.82 Å,同时MXene的厚度也从F终端的7.22 Å增加到I终端的8.49 Å。
此外,卤素吸附位点的对称性(Type I八面体、Type II三角棱柱)对稳定性有重要影响,Type I结构因更稳定的d轨道分裂构型而表现出更高的热力学稳定性。
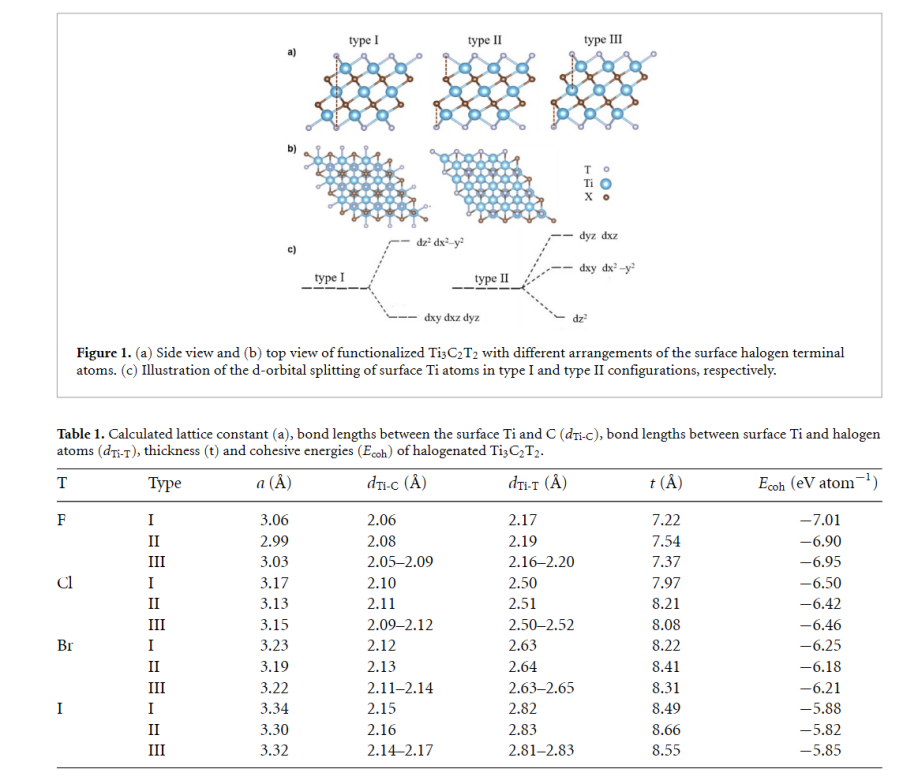
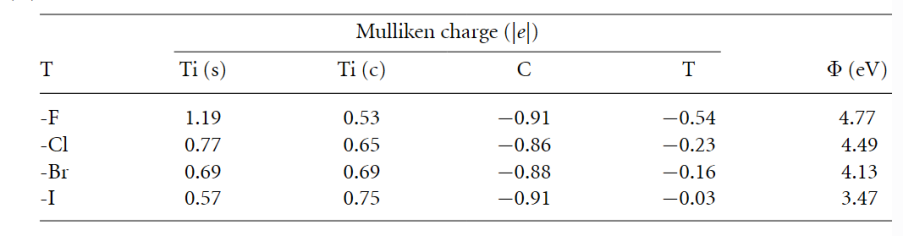
在电子性质方面,所有卤素终端的Ti₃C₂ MXene均呈现金属性,其费米能级附近的电子态主要由Ti 3d轨道贡献。然而,卤素的电负性差异导致键合特性显著不同:F终端因高电负性(4.0)与Ti形成强离子键,电荷转移明显,而Cl、Br、I终端则表现出更强的共价性,尤其是Ti–Cl键的p-d杂化显著。
这种差异进一步影响了材料的功函数,F终端的功函数最高(4.77 eV),而I终端最低(3.47 eV),表明后者具有更高的化学反应活性。
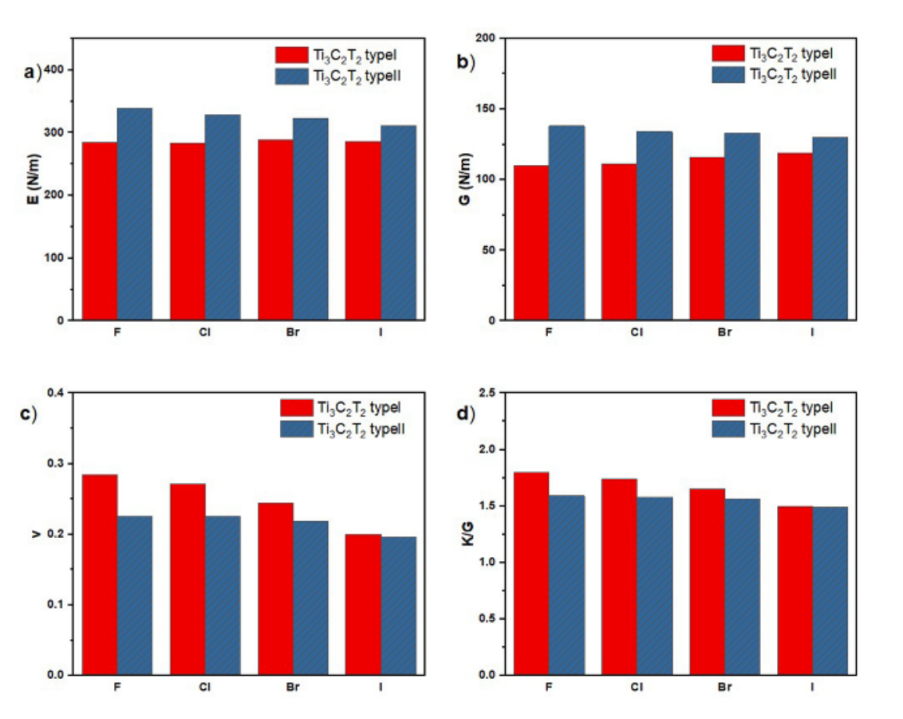
机械性能方面,卤素终端和吸附位点共同决定了MXene的力学行为。Type II结构因三角棱柱对称性而表现出更高的弹性模量,例如Type II Ti₃C₂F₂的Young’s模量达339 N/m。
同时,键合特性对材料的延展性影响显著:F终端因离子键的非方向性而表现出优异的延展性(K/G比值为1.80),而I终端因共价键主导而更脆。此外,Poisson比的变化表明I终端的柔性较差,限制了其在柔性器件中的应用潜力。
从应用角度来看,不同卤素终端的Ti₃C₂ MXene各具优势。F终端的高稳定性和离子特性使其适合电池电极材料,而Cl、Br、I终端因其共价性和高反应性,在催化、传感器及表面功能化(如Br终端可衍生出S、Se等新型终端)等领域更具潜力。
例如,实验研究表明,Br终端的Ti₃C₂ MXene可作为高效阴极材料用于锌离子电池,其放电容量显著高于Cl终端材料。
氧 / 羟基终端(-O, -OH)
-O, -OH的MXene结构在原子尺度上表现出独特的键合特征和电子性质。在MXene表面,终端基团(-O或-OH)与过渡金属(如Ti、Mo等)形成强键合,其中氧终端(-O)通常以桥接或端接方式与金属原子结合,键长约为1.8–2.1 Å(如Ti-O键约1.9 Å),而羟基(-OH)则以单齿配位(M-OH)形式存在,M-OH键长略长(如Ti-OH约2.0–2.2 Å)。
由于氧的电负性较高,-O终端会显著局域化MXene的电子分布,使金属d轨道部分氧化,导致费米能级附近态密度(DOS)降低,表现出半导体或窄带隙半金属特性。
相比之下,-OH终端的极化效应较弱,但仍会引入额外的电子态,特别是在价带顶附近形成OH-related杂化轨道,影响载流子迁移率。此外,-O和-OH终端共存时可能形成氢键网络(O…H-O),进一步调节层间相互作用,使MXene的层间距扩大至10–14 Å,并影响其机械性能和热稳定性。
从电子结构来看,氧终端MXene(如Ti₃C₂O₂)的计算研究表明,其导带主要由过渡金属d轨道贡献,而价带则包含金属d与O 2p轨道的杂化态,导致带隙可调(0.1–1.0 eV),具体取决于金属种类和终端覆盖率。
羟基终端的引入(如Ti₃C₂(OH)₂)会引入额外的H1s轨道,在费米能级附近形成浅能级缺陷态,可能增强载流子浓度但降低迁移率。此外,终端基团的排列方式(如有序或无序覆盖)也会影响MXene的电子性质,例如完全氧化的表面可能呈现更高的各向异性导电行为。
总体而言,氧/羟基终端通过改变MXene的键合构型、电荷分布和能带结构,使其展现出与传统二维材料不同的物理化学特性,为调控其电学、光学和磁学性能提供了重要手段。
硫族终端(-S, -Se, -Te)
硫族终端的 S、Se、Te 原子属于同一主族,电负性依次降低,原子半径逐渐增大。与卤素终端类似,电负性和原子半径的变化导致其与 MXene 基体的相互作用不同。S 原子的电负性相对较高,与MXene形成的键具有一定的离子性,而Se 和Te 原子与MXene 的键合逐渐向共价性转变。
在电子结构上,硫族终端会改变 MXene 表面的能带结构,随着原子序数增大,表面态密度在费米能级附近的分布发生变化,导电性能也会有所不同。
表面能方面,由于原子半径增大,表面能逐渐降低。化学活性上,硫族终端的 MXene 在吸附重金属离子或有机分子时表现出不同的亲和力,例如 – S 终端可能对某些过渡金属离子具有较强的吸附能力,而 – Te 终端可能因较大的原子半径,在分子识别中具有独特的选择性。
H终端的MXene在结构上保留了MXene的典型层状特征,但其表面化学性质因H键的引入而显著改变。与常见的-O或-F终端相比,H终端MXene展现出更低的电负性和更高的化学稳定性,其层间距通常小于含氧终端(约0.4-0.6 nm),这源于H原子较小的原子半径和较弱的层间相互作用。
理论计算表明,H终端MXene的电子结构呈现金属性或窄带隙半导体特性,费米能级附近存在显著的态密度,这为其在导电、催化等领域的应用奠定了基础。
热门MXene计算方法在MS&VASP催化计算课中均有讲解。