在科研实践中,基于量子力学 第一性原理计算 以其从头出发、不依赖实验参数的优势崭露头角,却也常因方法近似和计算成本而遭受质疑。 人们不禁要问:在面对复杂材料体系与多尺度过程时,它能否真正做到“所见即所得”? 本文将从理论基础 、泛函选择 、基组完备性 及与实验对比 等多个维度,系统剖析第一性原理计算 ,带您深入了解为何这一理论工具能够在材料设计与反应机理研究中屡次验证其预测价值。
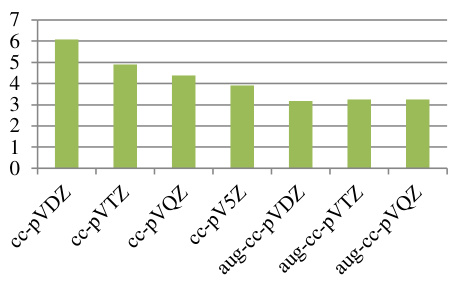
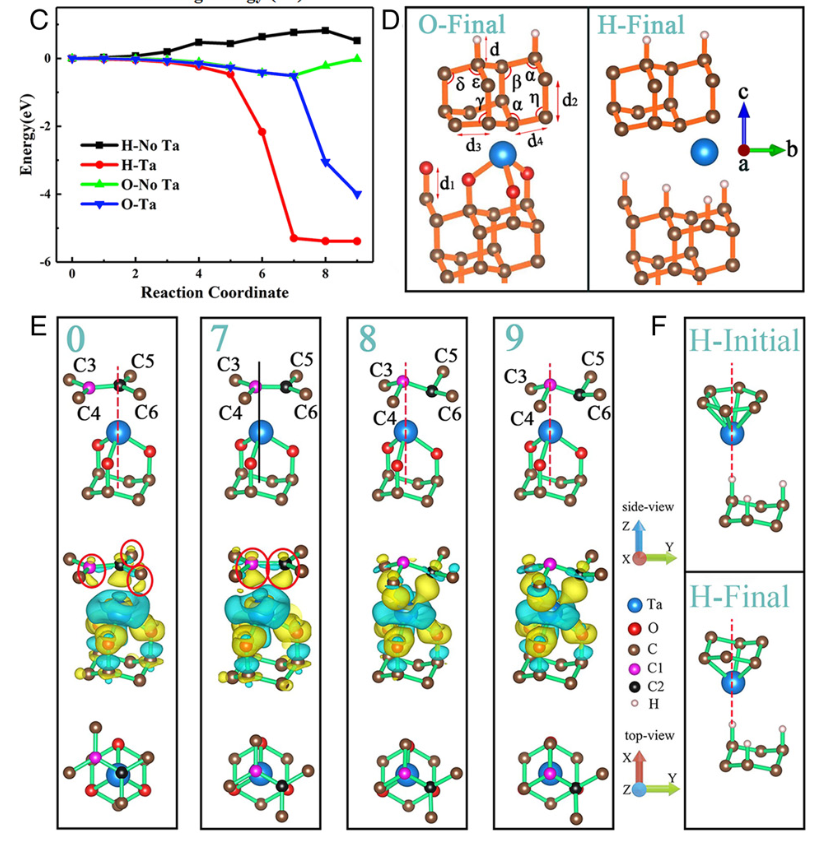
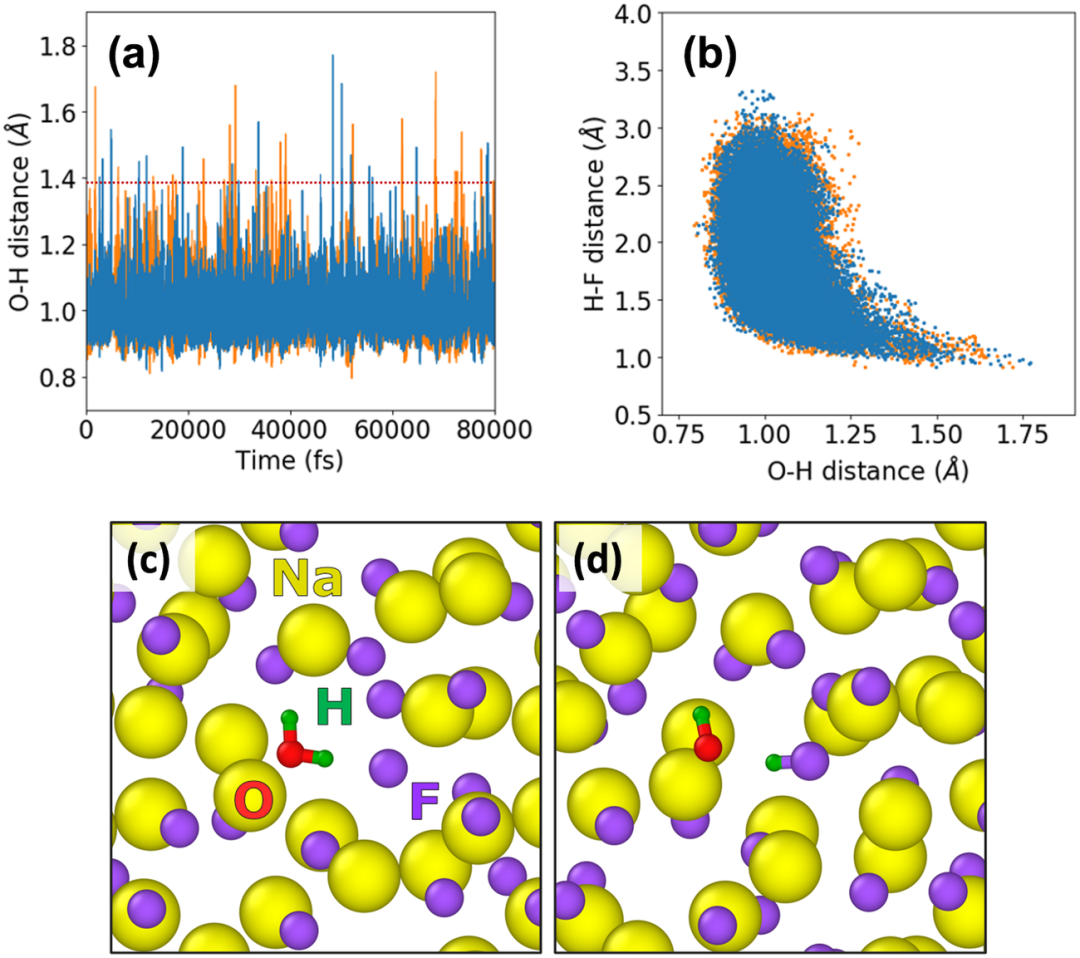
第一性原理 求解多粒子薛定谔方程 。根据Hohenberg-Kohn定理,基态电子密度唯一确定系统的所有性质 。Kohn-Sham方程通过引入虚拟的非相互作用电子体系,将多体问题简化为单电子方程求解,其中交换-关联泛函(XC泛函)的精确性直接影响结果精度 。例如,局域密度近似(LDA)和广义梯度近似(GGA)通过不同方式近似XC泛函,其误差通常控制在5%以内。 以石墨烯的电子结构计算为例,研究者利用平面波基组和GGA-PBE泛函优化晶格参数,得到与实验值偏差仅0.02 Å的晶格常数(理论值2.46 Å vs 实验值2.48 Å)。 这种高精度源于量子力学方程对电子间库仑排斥、动能等作用的精确描述,避免了经验参数引入的系统性偏差 。https://doi.org/10.1103/PhysRevB.78.205423 基组 作为波函数的数学展开基底,其完备性直接决定计算精度 。下表的对比数据显示:对于H₂二聚体结合能计算,使用aug-cc-pVQZ基组(含极化与弥散函数)时绝对误差仅为0.3 eV,而cc-pVDZ基组的误差高达2.1 eV。这是因为高阶基组能更精确描述电子云分布,特别是价电子的离域特性 。如下图所示,当基组从双ζ(DZ)扩展至五ζ(5Z)时,结合能逐渐收敛至理论极限值,弥散函数的加入(aug系列)进一步修正了长程相互作用。 DOI: 10.19113/sdufbed.76392 计算参数的收敛性设置是确保结果可靠性的关键 。以平面波截断能(Cutoff Energy)为例,中通过迭代正则化方法证明:当能量收敛阈值设为1 meV/atom时,晶格常数的波动范围可控制在0.01 Å以内。对于周期性体系,k点网格密度也需满足Brillouin区积分精度要求 。例如,在的锂离子电池模拟中,采用8×8×8 Monkhorst-Pack网格可使总能量误差小于0.1 meV/atom。传统DFT在处理强关联体系时存在局限性,而GW近似和动态平均场理论(DMFT)显著提升了计算精度 。显示,GW方法将氧化铜的带隙预测误差从DFT的1.2 eV降低至0.3 eV。通过对比了LDA、GGA和GW对CuO电子态密度的计算结果,GW修正后的价带顶与导带底位置更接近光电子能谱实验数据。 理论计算 实验部分通过短期生长实验和高分辨率透射电子显微镜(HRTEM)观察,验证了在CVD过程中,垂直生长的石墨烯片(VGs)逐渐演变为石墨纳米棒,并最终转变为金刚石,且发现Ta原子在薄膜中呈原子级分散,从而证实了理论计算的预测,即金刚石是通过石墨的相变形成的。
https://doi.org/10.1073/pnas.2201451119
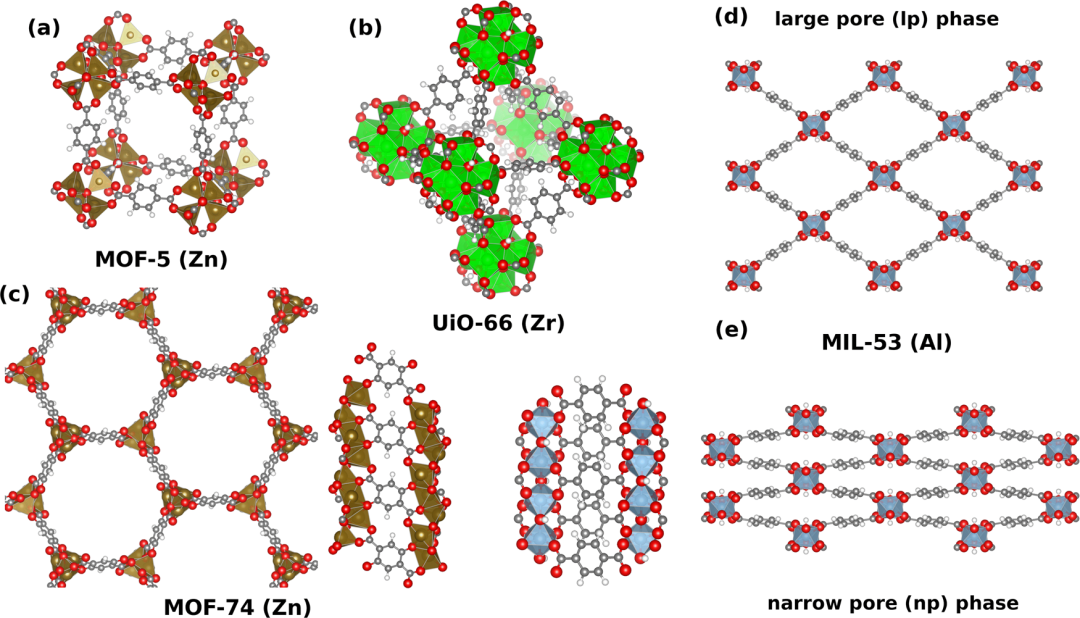
近年来,机器学习 (ML)与第一性原理的结合大幅提升了计算效率与精度 针对 MOF-5、UiO-66、MOF-74 和 MIL-53 等多种典型 MOFs,系统地研究其结构和动态性质,涵盖晶胞参数、能量、力、应力、弹性常数、热膨胀系数、热导率以及声子相关性质等多个方面。 研究人员致力于评估机器学习力场在描述这些性质时的准确性、计算效率以及与传统力场的性能差异,以提升 MOFs 计算建模水平。 https://doi.org/10.1038/s41524-024-01205-w 在氖高压熔化研究中,经典分子力学(CMD) 预测的熔化温度偏差达20%,而第一性原理分子动力学(AIMD) 的误差仅5%。这种差异源于CMD依赖经验势函数,无法精确描述电子云极化等量子效应。 有研究者运用从头算分子动力学(AIMD) 模拟研究了FeCr合金在含有H₂O的熔融NaF和NaCl盐中的腐蚀机制,发现 H₂O在两种熔盐中都高度稳定,解离成OH⁻和H⁺的情况很少见,并且这种解离是可逆的。 H₂O解离倾向与卤素原子的电负性呈正相关,与卤素原子的大小呈负相关 。H₂O以分子形式到达盐/金属界面,然后与金属发生反应。在盐/金属界面处,H₂O的反应从H⁺还原开始,而没有同时发生特定金属原子(如Cr)的氧化,表明还原和氧化是分步进行的,而不是通常认为的同时进行。还原的H原子更倾向于停留在界面处,并可能再次进入NaF盐,但在NaCl盐中不会。在NaF盐中,H₂O的部分解离更频繁,并伴随HF的形成。而在NaCl盐中,H₂O的解离较少,且没有观察到HCl的形成。在NaF盐中,还原的H原子主要沿盐/金属界面扩散;在NaCl盐中,还原的H原子更有可能进入金属区域。 https://doi.org/10.1038/s43246-024-00528-x 第一性原理计算的准确性 建立在量子力学严格性 、数值算法优化 与实验验证 的三重基础之上。随着基组扩展技术、多体修正方法和机器学习融合的发展,其预测精度已可达到化学精度(~1 kcal/mol)甚至更高 。未来,通过发展更精确的XC泛函 (如双杂化泛函 )和量子计算加速 ,第一性原理将在新材料设计 、催化机理解析 等领域发挥更关键的作用。 课程采用 Materials Studio CASTEP/DMol3 模块 作为主要引擎进行结构优化与性质计算,其中穿插密度泛函DFT基本理论 与晶体学基础知识 讲解,以及计算中所有参数的物理意义及设置方法。以二维材料的电子性质计算 为例,重点讲解科学问题的解决思路。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!