

 “从头算”(Ab initio)和“第一性原理”(First-principles)是理论计算科学中两个密切相关但又略有区别的概念,均指不依赖实验参数、从最基本的物理定律出发来研究材料或分子的性质。
“从头算”(Ab initio)和“第一性原理”(First-principles)是理论计算科学中两个密切相关但又略有区别的概念,均指不依赖实验参数、从最基本的物理定律出发来研究材料或分子的性质。“从头算”强调的是计算方法的理论纯净性,即完全从量子力学出发,不引入任何经验参数,代表性方法包括Hartree-Fock、配置相互作用(CI)和耦合簇(CC)等。
这些方法在精度上通常非常高,但计算代价大,常被用于小分子或简单体系的高精度研究。
而“第一性原理”更强调方法的普适性和系统性,其核心在于从基本物理定律(如薛定谔方程)出发建立通用模型,最典型的代表是密度泛函理论(DFT)。
DFT通过引入合理的泛函近似,显著提升了计算效率,使其能够广泛应用于固体物理、材料科学和纳米技术等领域。
因此,从头算更突出理论纯粹性,第一性原理则强调实用性与普适性,两者共同构成了现代计算材料科学的重要基础。
定义与核心思想
1.从头算(Ab Initio)
“从头算”(Ab Initio)是一类严格基于量子力学第一原理的计算方法,其名称源于拉丁语,意为“从一开始”,强调其理论体系不依赖任何实验数据或经验参数。
该类方法通过直接求解多体薛定谔方程来描述电子结构行为,从而预测材料性质或化学反应路径,具有高度的普适性和理论准确性。
典型的从头算流程包括三大步骤:首先,根据具体体系构建包含所有电子与原子核相互作用的多体薛定谔方程;
其次,采用一系列近似方法(如Hartree-Fock方法或密度泛函理论 DFT)将原本难以求解的多体问题简化为一组单电子方程;
最后,运用自洽场(SCF)迭代算法对电子密度进行反复更新,直至满足能量和密度的收敛条件。
虽然从头算计算代价较高,尤其在大体系或强相关电子问题中计算资源消耗显著,但其无需拟合参数、可预测性强等特点使其在催化材料、电子器件设计以及分子反应动力学研究中发挥着重要作用。
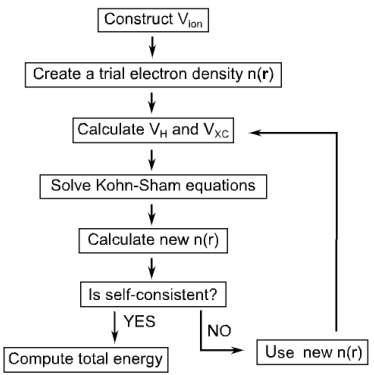
例如,Hartree-Fock方法通过假设单电子波函数的Slater行列式形式,避免了全电子波函数的维度灾难,但未充分处理电子关联效应。
2.第一性原理(First-Principles)
第一性原理(First-Principles)计算是一种从最基本的物理定律出发,不借助任何经验参数或拟合数据的理论建模方法。
在广义层面,它等同于从头算(Ab Initio),强调所有物理量的推导均源自如薛定谔方程或牛顿运动定律等基础方程。
然而,在材料科学、凝聚态物理和计算化学等具体应用中,第一性原理常狭义地指基于密度泛函理论(Density Functional Theory, DFT)的计算框架。
该方法以电子密度而非多体波函数为核心变量,通过求解Kohn-Sham方程来获得体系的基态能量和电子分布。
在实际求解中,必须引入交换–关联泛函(如LDA或GGA)以近似描述复杂的电子相关与交换效应,这也是DFT精度的关键控制因素。
第一性原理方法不仅可以预测材料的晶体结构、弹性常数、电子结构和光学响应,还在表界面化学、缺陷态分析以及催化机理研究中发挥重要作用。
其无需实验输入、适用于新材料探索的特性,使其成为现代计算材料设计的重要支撑手段。
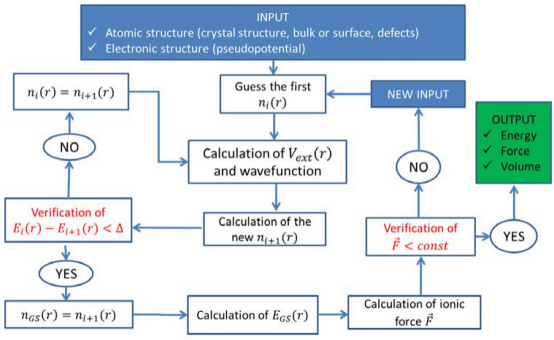
例如,DFT通过将多电子问题转化为单电子有效势,显著降低了计算复杂度,成为材料模拟的主流方法。
方法学对比与关联性
1.理论基础的重叠性
从头算(Ab Initio)和第一性原理(First-Principles)计算在理论基础上具有高度重叠性,均严格遵循量子力学的基本原理,主要依据多体薛定谔方程来描述粒子间的相互作用。
然而,两者的应用范围和技术细节存在差异。从头算在量子化学领域使用更为广泛,包含多种高精度电子结构方法,如Hartree-Fock(HF)、组态相互作用(CI)、耦合簇(CC)等,主要用于分子体系、非周期体系及小分子反应动力学等场景,强调电子相关的系统性处理。
相比之下,第一性原理计算在材料科学中更常与密度泛函理论(DFT)结合使用,特别适用于处理具有周期性边界条件的固体、表面或低维材料体系。
其最大优势在于计算效率高、可扩展性强,适合模拟大型材料单元和复杂构型。
因此,有文献指出,从头算是第一性原理方法的广义范畴之一,而第一性原理在狭义上往往特指DFT计算。
这种定义上的差异反映了学科领域在方法论选用上的倾向性与发展重点。
2.近似程度的差异
从头算方法与第一性原理(尤其是DFT)在处理电子结构问题时的理论框架存在显著差异。
从头算方法通常保留更严格的量子力学形式,诸如Hartree-Fock虽然在本质上忽略了部分电子关联效应,但其仍是基于精确的多电子波函数展开,强调从基本粒子态出发逐步构建整个体系的量子态。
这类方法的优点在于体系描述更具物理透明度,适用于需要高精度的化学反应路径、小分子势能面等研究。
相较而言,第一性原理计算,尤其以密度泛函理论(DFT)为代表,采用的是以电子密度为基本变量的近似框架,其核心在于交换–关联泛函的选择与构建,常用形式包括局域密度近似(LDA)和广义梯度近似(GGA)。
尽管DFT具备出色的计算效率,适合大尺度材料模拟,但在处理强关联体系(如过渡金属或强自旋–轨道耦合材料)时存在局限。
例如在铁基超导体FeSe的研究中,标准DFT方法无法准确再现实验中观测到的能带结构和磁性行为,必须引入如动态平均场理论(DMFT)等多体修正,才能更好地描述其d轨道电子的局域化特性。
而传统从头算方法虽然理论上更精确,但受限于指数级增长的计算资源需求,难以直接应用于这类复杂材料体系。
这一比较反映了方法选择需在理论完备性与计算可行性之间权衡,依据具体研究目标灵活采用。
3.计算流程的共性
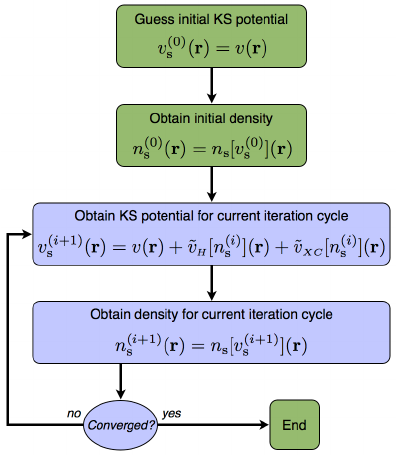
从头算方法与第一性原理计算尽管在理论基础与适用范围上存在差异,但它们在计算流程上具有高度一致性,均遵循自洽迭代的核心计算框架。
该流程通常包括以下几个关键步骤:首先,输入体系中原子的空间分布与初始电子密度;接着,根据电子密度构建总哈密顿量,其中包含离子势、电子间的Hartree库仑相互作用以及交换–关联势等项;
随后,通过求解Kohn-Sham方程(或在从头算中对应的多体薛定谔方程)获得更新的单电子波函数或密度;
最后,判断当前电子密度是否达到收敛标准,若未满足,则重新计算哈密顿量并继续迭代。
该循环过程确保电子密度与势场达到自洽状态,从而准确描述体系的基态性质。在实际应用中,为提高计算效率和适应不同体系的需求,通常需选择适当的数学工具与近似策略,例如在第一性原理计算中常使用平面波基组与赝势技术处理周期性固体,而在从头算中则可能采用高精度的原子轨道基组处理小分子体系。
通过这种结构化、系统性的流程,相关计算能够从量子力学基本原理出发,提供对材料和分子电子行为的可靠预测。
应用领域与典型案例
1.从头算的典型应用
从头算方法因其高精度和严谨的量子力学基础,被广泛应用于分子体系中复杂现象的理论研究,典型应用包括化学反应动力学与分子光谱学等领域。
在化学反应动力学研究中,从头算可用于构建准确的势能面,揭示反应路径与过渡态特征。
例如,针对甲硫醇(CH₃SH)与氢自由基(H·)之间的反应,研究人员采用双水平直接动力学方法,分别使用高精度的量子化学方法计算反应路径上的关键点,然后求解速率常数,并进一步比较产物分支比与实验结果,实现对反应动力学行为的定量预测与验证。
在分子光谱学领域,从头算方法同样发挥重要作用,尤其是在处理稀土元素等多电子体系时,Hartree-Fock方法结合组态相互作用(CI)可有效描述激发态结构。
例如在Ce³⁺或Eu²⁺等稀土离子的研究中,通过模拟其4f→5d跃迁的激发光谱,不仅有助于理解光致发光机制,还为发光材料设计提供理论依据。
这些应用充分体现了从头算方法在精确预测微观过程中的独特优势,尤其在无法依赖实验参数的体系中具有不可替代的价值。
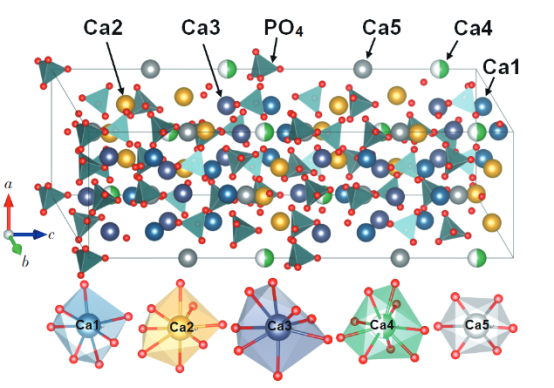
(DOI:10. 37188/CJL. 20220247)
2.第一性原理的突破性进展
第一性原理的突破性进展在多个领域中取得了显著的成果,为材料科学和物理学研究提供了新的视角和工具。
具体来说,在材料力学性质方面,利用密度泛函理论(DFT)计算了硅纳米线的弹性常数,包括杨氏模量和泊松比。
通过与纳米力学理论的预测结果进行对比,研究揭示了尺寸效应对这些力学性质的显著影响,这为理解和设计纳米尺度材料的力学行为提供了理论依据和计算支持。
在热电材料优化方面,结合电子–声子散射参数,研究预测了拉伸SiGe合金的Seebeck系数和热电优值ZT的变化趋势,并为实验合成提供了重要指导。
这一研究不仅为高效热电材料的设计提供了理论支持,也推动了热电能量转换技术的发展。
在超导体设计领域,研究深入探讨了铁基超导体的电子结构,揭示了磁序缺失与费米面缩小之间的矛盾。
通过这些研究,推动了多体修正模型的发展,进一步促进了超导体材料的理论研究和应用探索。
这些突破性进展不仅推动了基础研究的深入,也为新型材料的设计与应用开辟了新的道路。
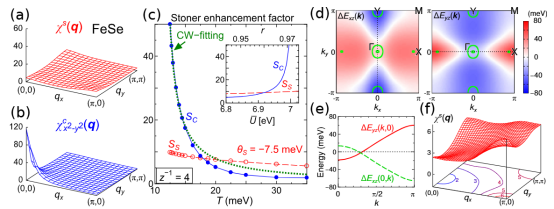
(DOI: https://doi.org/10.1103/PhysRevB.96.144509)
关联与区别的再探讨
1.哲学层面的差异
“从头算”和“第一性原理”在哲学层面上的差异主要体现在其对研究的纯粹性和普适性的侧重上。
具体来说,“从头算”更强调“从零开始”的思维方式,着重于从最基本的量子力学原理出发,进行全面的电子结构计算。
这种方法试图通过全电子计算来模拟系统的行为,无需依赖实验数据或外部假设,因此具备较强的理论纯粹性。
例如,量子化学中的全电子计算方法就是基于这一理念,其目标是从最基本的量子力学规律推导出系统的性质。
而“第一性原理”则更多强调“基于公理”的普适性,它依赖于已知的公理或基本定律,推导出系统的行为,具有更广泛的适用性和普适性。
例如,牛顿定律在经典力学中的作用就是一种基于公理的普适性原则,它为各种力学问题的解决提供了基础框架。
在这个意义上,埃隆·马斯克所提倡的“第一性原理思维”便源自于这一哲学理念,他认为问题应该被分解到最基本的不可再分单元,从而重新审视问题的本质,突破常规的思维束缚。
2.学科语境的分野
在不同学科的语境中,“从头算”和“第一性原理”有着显著的区别。
在物理学领域,两者的使用往往是互换的,都指代不依赖经验的量子力学计算方法。
具体来说,物理学家通常将这两者视为基于量子力学原理、从基本粒子层面推导物质性质的方法,进而进行无经验假设的计算。
然而,在化学领域,“从头算”通常指的是特定的计算方法,如Hartree-Fock方法及其更高阶的扩展(如密度泛函理论DFT)。
这些方法侧重于解决电子结构问题,并且注重计算的精确性。
而“第一性原理”在化学领域更多地指代基于密度泛函理论(DFT)的计算方法,这种方法通过近似处理电子交换和相关效应,在保证计算效率的同时,能够提供较为准确的物质性质预测。
不同学科对这些术语的定义差异,要求跨学科的研究者在合作中明确术语的具体含义,以避免概念上的混淆和理解上的差异。这也反映了不同学科对于计算方法的不同关注点和需求。
3.计算效率与精度的权衡
在实际应用中,“从头算”和“第一性原理”之间常常存在着计算效率与精度的权衡问题。
以“从头算”中的配置相互作用(CI)方法为例,虽然这种方法能够提供极高的计算精度,特别是在处理分子体系时表现出色,但其计算量随着电子数目的增加呈指数级增长,因此对于较大体系的计算会变得非常困难,通常只能应用于小分子或简单体系。
与此不同,第一性原理方法,尤其是基于密度泛函理论(DFT)的方法,通过采用泛函近似和其他计算技巧,能够大大提高计算效率,使得处理更大规模的体系成为可能。
具体来说,DFT的计算复杂度通常为O(N³),这意味着它能够处理从几十到几千个原子的体系,极大地拓宽了其应用范围。
尽管DFT相较于CI方法在精度上有所牺牲,但其在处理大规模体系时的优势,使得它在材料科学、化学反应等领域得到了广泛应用。
因此,研究人员往往需要在精度和计算效率之间做出选择,以适应不同的研究需求和计算资源。
(https://doi.org/10.3390/batteries9020086)
以VASP软件为例,其平面波基组和赝势技术使DFT计算效率提升10倍以上,推动了高通量材料筛选。
未来挑战与多尺度融合
1.强关联体系的瓶颈
在强关联电子体系中,如铁基超导体和重费米子材料,传统的密度泛函理论(DFT)往往无法提供准确的电子结构描述。
这主要源于DFT中交换–关联泛函(如LDA或GGA)在处理强电子–电子相互作用时的局限性,特别是在d或f轨道占据主导地位的材料中。
这些体系的物理行为受到局域电子关联的显著影响,导致传统方法低估能隙、错误预测磁性或失去金属–绝缘转变的关键特征。
因此,需要引入更复杂的理论方法,如动态平均场理论(DMFT)或量子蒙特卡洛模拟,以更真实地刻画电子的局域化行为及其动力学特性。
这类方法虽计算成本较高,但在描述强关联体系的金属–绝缘转变、超导机制及非费米液行为等方面具有显著优势。
2.机器学习辅助的机遇
随着数据驱动科学的兴起,机器学习在从头算计算中展现出巨大潜力。
以AIGLE框架为例,该方法通过构建机器学习模型来近似复杂的记忆核函数,从而显著降低多体动力学计算的时间成本。
传统从头算模拟在处理大体系或长时间演化过程中,受限于计算资源,难以实现跨尺度的物理建模。
而AIGLE等新方法通过“学习”量子系统的动力学行为,能够在保留高精度的基础上大幅提高计算效率,最终将量子力学模拟拓展至介观尺度。
这类融合策略不仅有助于解决高维计算中的“维度灾难”,也为多物理场耦合与复杂系统建模提供了新的技术路径,正成为当前材料设计与电子结构研究的重要发展方向。
3.实验–计算闭环验证
在材料科学研究中,实验与理论的深度融合正成为提升预测可靠性与加速新材料发现的关键路径。
以同步辐射X射线衍射(XRD)为代表的高通量实验技术,能够快速获得材料结构演化信息。
而理论方面,通过多尺度模拟方法(如DFT结合分子动力学),可以构建出与实验条件高度一致的模拟体系。
两者结合形成“闭环验证”机制,即在模拟预测指导下进行实验筛选,实验结果反过来用于修正和优化理论模型。
例如,通过对比实验获得的晶体结构或电子态密度与DFT结果,可以校准交换–关联泛函或赝势选择,从而显著提升理论模型的可信度。
这一机制在电催化、热电材料与新型二维材料研究中已获得广泛应用,并正逐步向自动化与智能化设计平台演进。
结语
从头算与第一性原理作为量子力学的双翼,共同构筑了现代计算科学的基石。
它们在定义上的微妙差异反映了学科发展的历史路径,而方法学的融合(如DFT作为第一性原理的核心)则彰显了理论应用的普适性。
未来,随着算法优化与跨尺度建模的突破,两者将在纳米材料、能源存储及量子器件等领域持续释放创新潜力。