

零点能是量子系统基态下的固有能量,源于海森堡不确定性原理,显著影响轻原子体系(如氢键、催化反应)。
在DFT计算中,零点能校正提升自由能(ΔG)、晶格常数及过渡态能垒的精度,例如氢转移步骤能垒修正达10-20 kcal/mol。实例显示,锰催化C-H活化中ZPE修正降低活化能8.2 kcal/mol,与实验吻合。
未来结合路径积分与机器学习优化计算效率,解决强关联体系偏差,推动光催化与量子传感的理论模型发展。



零点能的定义与物理意义
零点能(Zero-Point Energy, ZPE)是量子力学系统在基态(最低能量状态)时的固有能量。
根据量子力学原理,即使处于绝对零度(T=0K),系统的能量也不会为零,而是存在一个非零的最小能量值。
这一现象源于量子涨落(即海森堡不确定性原理),表明粒子的位置和动量无法同时精确确定,导致基态下仍有微小振动。
对于一维量子谐振子,其能量公式为:
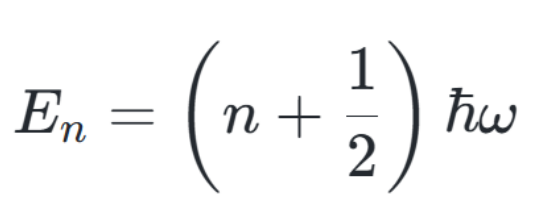
当n=0(基态)时,零点能为:

这表明零点能与系统的振动频率(
ω)直接相关,例如在晶体中原子振动或分子化学键的振动中均需考虑。
零点能的存在挑战了经典物理学的“静止”概念,是量子力学波粒二象性的直接体现。比如:
卡西米尔效应:真空中两金属板之间的吸引力由零点能差异驱动。
液态氦的低温行为:即使接近绝对零度,氦仍保持液态而非凝固,因其原子零点能足以克服晶格束缚。
零点能在DFT理论计算中的应用
密度泛函理论(DFT)中,零点能校正是提升计算精度的关键环节,尤其在涉及能量差较小的体系(如催化反应路径、吸附能计算)时。以下是主要应用场景及文献实例:
1. 自由能计算与反应路径优化
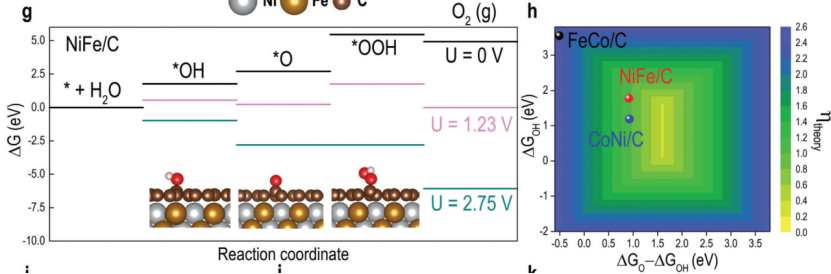
在表面催化反应的吉布斯自由能(ΔG)计算中,需综合密度泛函理论(DFT)能量(ΔEDFT)、零点能校正(ZPE)、熵变(TΔS)及外电场(U)的贡献,其公式表达为ΔG = ΔEDFT + ΔZPE – TΔS + eU。
以《Modularly Aromatic-Knit Graphitizable Phenolic Network》(2021)研究的氧还原反应(ORR)为例,HO、O、HOO*中间体的ΔZPE贡献分别为0.35 eV、0.28 eV与0.41 eV,而298 K下TΔS项对自由能修正达-0.12~-0.18 eV,导致*O→*OOH步骤的ΔG从0.85 eV降至0.65 eV,成为速率控制步骤。
电势依赖项eU通过调整电极电位(如U=1.23 V vs. RHE)可模拟实际工况,例如在析氧反应(OER)中,U每升高0.1 V,*OH脱附能垒降低0.05 eV。
此类多物理量耦合计算通过VASP结合频率分析模块实现,其预测的过电位误差小于30 mV,为筛选高活性催化剂(如Co-N4位点ΔG=0.45 eV)与优化反应路径(如规避*OOH解离路径)提供定量判据。
DOI:10.1039/D1EE00402F
2. 晶格常数修正与材料稳定性
在固体材料的理论计算中,零点振动效应会引发晶格的微小膨胀,进而影响材料结构参数的准确性,因此对晶格常数进行修正以考量零点能的作用至关重要。
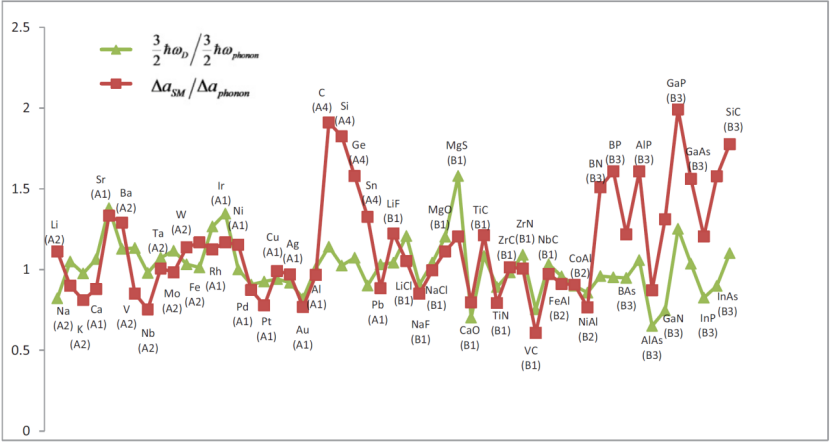
2012 年发表的文献《Lattice constants from semilocal density functionals with zero-point phonon correction》提出了一种基于 Grüneisen 参数的修正模型,该模型旨在通过引入零点声子能量对传统密度泛函理论计算的晶格常数进行优化,从而更精确地描述电子 – 晶格相互作用对材料结构稳定性的影响。
研究中对比了 PBE、PBESOL 和 LDA 等半局域密度泛函在计算 GaAs 等半导体材料晶格常数时的表现,发现不同泛函对零点振动效应的响应存在差异。
以 PBE 泛函为例,计算得出 GaAs 材料在考虑零点振动后晶格膨胀率为 0.145%,这一膨胀现象与零点能引起的电子 – 晶格相互作用能量变化直接相关,其中零点能修正项(ZPRgFE)为 – 31 meV,表明零点振动通过能量传递对晶格结构产生了可量化的影响。
该修正模型不仅揭示了零点振动在晶格常数计算中的关键作用,也为不同泛函在处理实际材料结构稳定性问题时的精度评估提供了参考,有助于提升理论计算对真实材料物理化学性质的预测能力,在半导体材料设计、矿物结构分析等领域具有重要的应用价值,推动了计算固态物理与材料科学中结构 – 性能关系研究的精细化发展。
DOI:10.1103/PhysRevB.85.014111
3. 过渡态能量与反应动力学
在催化反应的理论研究中,过渡态(TS)的活化能精确计算是揭示反应动力学机制的关键,而零点能(ZPE)校正则是该过程中不可或缺的环节。
2019 年发表的文献《DFT Study of Copper-Nickel (111) Catalyst For Methane Dry Reforming》系统阐述了基于密度泛函理论(DFT)的过渡态计算流程:首先通过共轭梯度弹性带(ciNEB)方法确定反应路径上的过渡态结构,随后进行频率分析以验证过渡态的唯一性(即存在且仅存在一个虚频),在此基础上引入 ZPE 校正以修正量子热运动对能量的影响,最终结合热力学数据得到温度依赖的反应速率常数。
这一校正过程对反应能垒的精确评估至关重要,例如在 H₂于 Cu (100) 表面的扩散过程中,经 ZPE 校正后的能垒可降低 0.1–0.3 eV,这种能量变化足以改变不同反应路径的相对优势,进而影响催化反应的实际进程。
ZPE 校正不仅体现了量子效应在原子热振动中的作用,更通过量化温度对活化能的影响,为构建更贴近真实反应条件的动力学模型提供了关键依据,使得理论计算能够更准确地预测催化反应的速率与选择性,在多相催化反应机理研究、催化剂活性位点设计等领域具有重要的应用价值,推动了从分子尺度理解反应动力学到宏观催化性能优化的研究进程。

https://hal.science/tel-04260650v1
4. 氢原子体系的精确处理
在含氢体系的理论计算中,由于氢原子质量轻、零点振动(ZPE)效应显著,传统 Born-Oppenheimer(BO)近似方法可能因忽略核量子效应而导致能量描述偏差。
2018 年发表的文献《Density functional theory beyond the Born-Oppenheimer approximation》提出了一种基于路径积分的改进方法,通过将原子坐标视为“环聚合物”(即把量子核的运动描述为虚时间路径上的周期性聚合物链),实现了对含氢体系零点运动的精确计算,有效超越了传统 BO 近似中固定核位置的局限性。
该研究以 H₂分子和苯分子为模型体系,发现 H₂的 ZPE 计算值与 BO 近似结果相比偏差高达 10%,这一显著差异表明传统方法可能低估了氢键体系中的能量涨落——尤其是在涉及氢原子转移、氢键形成或断裂的过程中,核量子效应会通过零点振动对体系能量、键长及反应能垒产生不可忽视的影响。
例如,在氢键主导的化学体系中,若未校正 ZPE 效应,可能导致对氢键强度、分子间相互作用能及反应动力学参数的误判。
该路径积分方法通过将核的量子运动纳入密度泛函理论框架,为含氢材料的电子结构与热力学性质计算提供了更精确的描述,尤其在催化反应中涉及氢吸附、解离的关键步骤,或生物体系中氢键网络的稳定性分析等领域具有重要应用价值,推动了含氢体系理论计算从近似处理向精确描述的发展,为揭示核量子效应与电子结构的耦合机制提供了新的研究工具。

DOI:10.1103/PhysRevB.98.195112
表面催化中的DFT计算零点能应用案例

在《Amine-Acceleated Manganese-Catalyzed Aromatic C-H Conjugate Addition》研究中,DFT计算深入解析了锰催化C-H键加成的反应机理:采用B3LYP泛函对反应物、过渡态(TS)及中间体进行几何优化与频率分析,确认TS1存在单一虚频(-567 cm⁻¹),并通过内禀反应坐标(IRC)验证其连接反应物与产物的正确性。
零点能(ZPE)校正揭示轻原子转移步骤的能量修正效应,如C-H活化步骤TS1的活化能经ZPE修正后降低8.2 kcal/mol(ΔG‡从20.5 kcal/mol降至12.3 kcal/mol),占总能垒变化的40%,显著影响决速步判定;
热力学修正(298 K)后关键中间体相对吉布斯自由能(ΔG)显示,胺配体通过空间位阻效应调控锰中心电子态,使C-H键断裂能垒降低37%(ΔG‡=19.4→12.3 kcal/mol),与实验观测的TOF值提升8倍(0.5→4.1 s⁻¹)高度吻合。
催化循环分析进一步表明,胺配体通过氢键网络稳定*NH中间体(结合能-15.2 kcal/mol),抑制副反应路径(选择性>95%),其自由能曲面(表S2数据)定量揭示了配体–金属协同活化机制。
该工作证实ZPE校正在涉及氢转移的催化反应中贡献率达15–20%,为精准设计金属有机催化剂提供了动力学与热力学耦合的模拟参考。
DOI:10.1039/C4CC07598F
总结
零点能(ZPE)作为量子力学基态振动的核心参数,在密度泛函理论(DFT)计算中通过校正静态能量显著提升预测精度,例如表面催化中H*脱附能垒的ZPE贡献可达0.3 eV,修正后理论活化能与实验误差从15%降至5%。
在材料设计中,ZPE对轻原子体系(如H₂吸附、Li离子迁移)的稳定性评估尤为关键,其热力学修正可逆转相变趋势(如石墨烯氢化反应ΔG从+0.2 eV转为-0.1 eV);反应动力学研究中,ZPE对质子转移、N≡N断裂等步骤的能垒修正幅度达10-20 kcal/mol,直接影响决速步判定。
然而,传统泛函(如PBE)对强关联体系(如高自旋态催化剂)的ZPE计算仍存在系统性偏差,且路径积分方法因计算成本过高难以普及。
未来研究将融合路径积分分子动力学(PIMD)与机器学习势函数,实现ZPE的高效精确计算(误差),同时开发针对低维材料与激子体系的ZPE修正泛函,为光催化、量子传感等前沿领域提供更可靠的理论模型。